Association Among Ccr5 Genotypes, Ccr5 Expression, and In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enhanced Monocyte Migration to CXCR3 and CCR5 Chemokines in COPD
ERJ Express. Published on March 10, 2016 as doi: 10.1183/13993003.01642-2015 ORIGINAL ARTICLE IN PRESS | CORRECTED PROOF Enhanced monocyte migration to CXCR3 and CCR5 chemokines in COPD Claudia Costa1, Suzanne L. Traves1, Susan J. Tudhope1, Peter S. Fenwick1, Kylie B.R. Belchamber1, Richard E.K. Russell2, Peter J. Barnes1 and Louise E. Donnelly1 Affiliations: 1Airway Disease, National Heart and Lung Institute, Imperial College London, London, UK. 2Chest Clinic, King Edward King VII Hospital, Windsor, UK. Correspondence: Louise E. Donnelly, Airway Disease, National Heart and Lung Institute, Dovehouse Street, London, SW3 6LY, UK. E-mail: [email protected] ABSTRACT Chronic obstructive pulmonary disease (COPD) patients exhibit chronic inflammation, both in the lung parenchyma and the airways, which is characterised by an increased infiltration of macrophages and T-lymphocytes, particularly CD8+ cells. Both cell types can express chemokine (C-X-C motif) receptor (CXCR)3 and C-C chemokine receptor 5 and the relevant chemokines for these receptors are elevated in COPD. The aim of this study was to compare chemotactic responses of lymphocytes and monocytes of nonsmokers, smokers and COPD patients towards CXCR3 ligands and chemokine (C-C motif) ligand (CCL)5. Migration of peripheral blood mononuclear cells, monocytes and lymphocytes from nonsmokers, smokers and COPD patients toward CXCR3 chemokines and CCL5 was analysed using chemotaxis assays. There was increased migration of peripheral blood mononuclear cells from COPD patients towards all chemokines studied when compared with nonsmokers and smokers. Both lymphocytes and monocytes contributed to this enhanced response, which was not explained by increased receptor expression. -

Human Th17 Cells Share Major Trafficking Receptors with Both Polarized Effector T Cells and FOXP3+ Regulatory T Cells
Human Th17 Cells Share Major Trafficking Receptors with Both Polarized Effector T Cells and FOXP3+ Regulatory T Cells This information is current as Hyung W. Lim, Jeeho Lee, Peter Hillsamer and Chang H. of September 28, 2021. Kim J Immunol 2008; 180:122-129; ; doi: 10.4049/jimmunol.180.1.122 http://www.jimmunol.org/content/180/1/122 Downloaded from References This article cites 44 articles, 15 of which you can access for free at: http://www.jimmunol.org/content/180/1/122.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2008 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Human Th17 Cells Share Major Trafficking Receptors with Both Polarized Effector T Cells and FOXP3؉ Regulatory T Cells1 Hyung W. Lim,* Jeeho Lee,* Peter Hillsamer,† and Chang H. Kim2* It is a question of interest whether Th17 cells express trafficking receptors unique to this Th cell lineage and migrate specifically to certain tissue sites. -
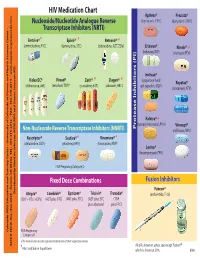
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (Tipranavir, TPV) (Darunavir, DRV) Transcriptase Inhibitors (NRTI)
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (tipranavir, TPV) (darunavir, DRV) Transcriptase Inhibitors (NRTI) Emtriva®* Epivir® * Retrovir® * (emtricitabine, FTC) (lamivudine, 3TC) (zidovudine, AZT, ZDV) Crixivan® Norvir® * (indinavir, IDV) (ritonavir, RTV) Invirase® Videx EC® Viread® Zerit® Ziagen® * * (saquinavir hard Reyataz® (didanosine, ddl) (tenofovir,TDF)* (stavudine, d4T) (abacavir, ABC) gel capsules, SQV) (atazanavir, ATV) Kaletra® * (lopinavir/ritonavir, LPV/r) Viracept® Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) Inhibitors (PI) Protease (nelfinavir, NFV) Rescriptor® Sustiva®* Viramune® * (delavirdine, DLV) (efavirenz, EFV) (nevirapine, NVP) Lexiva® (fosamprenavir, FPV) FDA Pregnancy Category D Fixed Dose Combinations Fusion Inhibitors Fuzeon® Developed by MeriLou Johnson, MeriLou by Developed Johnson, MSW, and Steven MPA MD Atripla® Combivir® Epzicom® Trizivir® Truvada® (enfuvirtide,T-20) (TDF+FTC+EFV) (AZT plus 3TC) (ABC plus 3TC) (AZT plus 3TC (TDF plus abacavir) plus FTC) Colorado AIDS Education and Training Center • University of Colorado at Denver and Health Sciences Center • Center and Health Sciences Denver at of Colorado • University Center Training and AIDS Education Colorado FDA Pregnancy Category D ®The brands listed are the registered trademarks of their respective owners. 4200 E. Ave., Ninth TEL: A089 • Denver, Box 80262 CO 303-315-2512 • FAX: • 303-315-2514 • www.mpaetc.org/colorado.htm All pills shown in actual size except Fuzeon® *Also available in liquid form. which is shown at 50%. 8/06 Medication Schedule Name______________________________ Date___________ Number of Time of day you ar to take TOTAL Name of Medication pills to take this medicine Food Interactions Side Effects number of each time pills each day With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ Discontinued Medications or Formulations Helpful Hints: • Refill prescriptions before you run out. -

Truvada (Emtricitabine / Tenofovir Disoproxil)
Pre-exposure Prophylaxis (2.3) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use Recommended dose in HIV-1 uninfected adults: One tablet TRUVADA safely and effectively. See full prescribing information (containing 200 mg/300 mg of emtricitabine and tenofovir for TRUVADA. disoproxil fumarate) once daily taken orally with or without food. (2.3) TRUVADA® (emtricitabine/tenofovir disoproxil fumarate) tablets, for oral use Recommended dose in renally impaired HIV-uninfected Initial U.S. Approval: 2004 individuals: Do not use TRUVADA in HIV-uninfected individuals if CrCl is below 60 mL/min. If a decrease in CrCl is observed in WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH uninfected individuals while using TRUVADA for PrEP, evaluate STEATOSIS, POST-TREATMENT ACUTE EXACERBATION OF potential causes and re-assess potential risks and benefits of HEPATITIS B, and RISK OF DRUG RESISTANCE WITH USE OF continued use. (2.4) TRUVADA FOR PrEP IN UNDIAGNOSED HIV-1 INFECTION -----------------------DOSAGE FORMS AND STRENGTHS------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 Lactic acidosis and severe hepatomegaly with steatosis, mg/150 mg of emtricitabine and tenofovir disoproxil fumarate . (3) including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, a component of TRUVADA. (5.1) --------------------------------CONTRAINDICATIONS----------------------------- TRUVADA is not approved for the treatment of chronic Do not use TRUVADA for pre-exposure prophylaxis in individuals with hepatitis B virus (HBV) infection. Severe acute unknown or positive HIV-1 status. TRUVADA should be used in exacerbations of hepatitis B have been reported in patients HIV-infected patients only in combination with other antiretroviral coinfected with HIV-1 and HBV who have discontinued agents. -

Download Article PDF/Slides
New Antiretrovirals in Development: Reprinted from The PRN Notebook,™ june 2002. Dr. James F. Braun, Editor-in-Chief. Tim Horn, Executive Editor. Published in New York City by the Physicians’ Research Network, Inc.,® John Graham Brown, Executive Director. For further information and other articles The View in 2002 available online, visit http://www.PRN.org All rights reserved. © june 2002. Roy “Trip” Gulick, md, mph Associate Professor of Medicine, Weill Medical College of Cornell University Director, Cornell Clinical Trials Unit, New York, New York Summary by Tim Horn Edited by Scott Hammer, md espite the fact that 16 antiretro- tiviral activity of emtricitabine was estab- Preliminary results from two random- virals are approved for use in the lished, with total daily doses of 200 mg or ized studies—FTC-302 and FTC-303—were United States, there is an indis- more producing the greatest median viral reported by Dr. Charles van der Horst and putable need for new anti-hiv com- load suppression: 1.72-1.92 log. Based on his colleagues at the 8th croi, held in Feb- pounds that have potent and these data, a once-daily dose of 200 mg ruary 2001 in Chicago (van der Horst, durable efficacy profiles, unique re- was selected for further long-term clinical 2001). FTC-302 was a blinded comparison sistance patterns, patient-friendly dosing study. “This is what we’re looking forward of emtricitabine and lamivudine, both in schedules, and minimal toxicities. To pro- to with emtricitabine,” commented Dr. combination with stavudine (Zerit) and vide prn with a glimpse of drugs current- Gulick. -

CCR5 Deficiency Impairs CD4+ T Cell Memory Responses and Antigenic Sensitivity
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.14.948893; this version posted February 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. CCR5 deficiency impairs CD4+ T cell memory responses and antigenic sensitivity through increased ceramide synthesis Ana Martín-Leal1§, Raquel Blanco1§, Josefina Casas2,3, María E. Sáez4, Elena Rodríguez- Bovolenta5, Itziar de Rojas6, Carina Drechsler7,8,9, Luis Miguel Real10,11, Gemma Fabrias2,3, Agustín Ruíz6,12, Mario Castro13, Wolfgang W.A. Schamel7,8,14, Balbino Alarcón5, Hisse M. van Santen5, Santos Mañes1* 1Department of Immunology and Oncology, Centro Nacional de Biotecnología (CNB/CSIC), Madrid, Spain; 2Department of Biological Chemistry, Institute of Advanced Chemistry of Catalonia (IQAC-CSIC), Barcelona, Spain and 3CIBER Liver and Digestive Diseases (CIBER-EDH), Instituto de Salud Carlos III, Madrid, Spain; 4Centro Andaluz de Estudios Bioinformáticos (CAEBi), Seville, Spain; 5Department of Cell Biology and Immunology, Centro de Biología Molecular Severo Ochoa (CBMSO/CSIC), Madrid, Spain; 6Alzheimer Research Center and Memory Clinic of the Fundació ACE, Institut Català de Neurociències Aplicades, Barcelona, Spain; 7Signaling Research Centers BIOSS and CIBSS, 8Department of Immunology, Faculty of Biology and 9Institute for Pharmaceutical Sciences, University of Freiburg, Freiburg, Germany; 10Unit of Infectious Diseases and Microbiology, Hospital Universitario de Valme, -

Product Monograph for CELSENTRI
PRODUCT MONOGRAPH PrCELSENTRI maraviroc Tablets 150 and 300 mg CCR5 antagonist ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Revision: July 05, 2019 Submission Control No: 226222 © 2019 ViiV Healthcare group of companies or its licensor. Trademarks are owned by or licensed to the ViiV Healthcare group of companies. Page 1 of 60 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION.........................................................3 SUMMARY PRODUCT INFORMATION ........................................................................3 INDICATIONS AND CLINICAL USE..............................................................................3 CONTRAINDICATIONS ...................................................................................................3 WARNINGS AND PRECAUTIONS..................................................................................4 ADVERSE REACTIONS....................................................................................................9 DRUG INTERACTIONS ..................................................................................................19 DOSAGE AND ADMINISTRATION..............................................................................28 OVERDOSAGE ................................................................................................................31 ACTION AND CLINICAL PHARMACOLOGY ............................................................31 STORAGE AND STABILITY..........................................................................................36 -

Reference ID: 2998411
• New onset or worsening renal impairment: Can include acute HIGHLIGHTS OF PRESCRIBING INFORMATION renal failure and Fanconi syndrome. Assess creatinine clearance These highlights do not include all the information needed to use (CrCl) before initiating treatment with COMPLERA. Monitor CrCl COMPLERA safely and effectively. See full prescribing and serum phosphorus in patients at risk. Avoid administering information for COMPLERA. COMPLERA with concurrent or recent use of nephrotoxic drugs. (5.3) COMPLERATM (emtricitabine/rilpivirine/tenofovir disoproxil • Caution should be given to prescribing COMPLERA with drugs fumarate) tablets that may reduce the exposure of rilpivirine. (5.4) Initial U.S. Approval: 2011 • Caution should be given to prescribing COMPLERA with drugs with a known risk of Torsade de Pointes. (5.4) WARNINGS: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS and POST TREATMENT ACUTE • Depressive disorders: Severe depressive disorders (depressed EXACERBATION OF HEPATITIS B mood, depression, dysphoria, major depression, mood altered, negative thoughts, suicide attempt, suicidal ideation) have been See full prescribing information for complete boxed warning. reported. Immediate medical evaluation is recommended for severe depressive disorders. (5.5) • Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of • Decreases in bone mineral density (BMD): Consider monitoring nucleoside analogs, including tenofovir disoproxil BMD in patients with a history of pathologic fracture -

AGENERASE® (Amprenavir) Capsules
NDA 21-007/SLR017 Page 2 PRESCRIBING INFORMATION AGENERASE® (amprenavir) Capsules PATIENT INFORMATION INCLUDED Because of the potential risk of toxicity from the large amount of the excipient, propylene glycol, contained in AGENERASE Oral Solution, that formulation is contraindicated in infants and children below the age of 4 years and certain other patient populations and should be used with caution in others. Consult the complete prescribing information for AGENERASE Oral Solution for full information. DESCRIPTION AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus (HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4- amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula: Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C. AGENERASE Capsules are available for oral administration. Each 50-mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50-mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU. -

CXCR6 Within T-Helper (Th) and T-Cytotoxic
European Journal of Endocrinology (2005) 152 635–643 ISSN 0804-4643 EXPERIMENTAL STUDY CXCR6 within T-helper (Th) and T-cytotoxic (Tc) type 1 lymphocytes in Graves’ disease (GD) G Aust, M Kamprad1, P Lamesch2 and E Schmu¨cking Institute of Anatomy, 1Department of Clinical Immunology and Transfusion Medicine and 2Department of Surgery, University of Leipzig, Phillipp-Rosenthal-Str. 55, Leipzig, 04103, Germany (Correspondence should be addressed to G Aust; Email: [email protected]) Abstract Objective: In Graves’ disease (GD), stimulating anti-TSH receptor antibodies are responsible for hyperthyroidism. T-helper 2 (Th2) cells were expected to be involved in the underlying immune mech- anism, although this is still controversial. The aim of this study was to examine the expression of CXCR6, a chemokine receptor that marks functionally specialized T-cells within the Th1 and T-cyto- toxic 1 (Tc1) cell pool, to gain new insights into the running immune processes. Methods: CXCR6 expression was examined on peripheral blood lymphocytes (PBLs) and thyroid- derived lymphocytes (TLs) of GD patients in flow cytometry. CXCR6 cDNA was quantified in thyroid tissues affected by GD (n ¼ 16), Hashimoto’s thyroiditis (HT; n ¼ 2) and thyroid autonomy (TA; n ¼ 11) using real-time reverse transcriptase PCR. Results: The percentages of peripheral CXCR6þ PBLs did not differ between GD and normal subjects. CXCR6 was expressed by small subsets of circulating T-cells and natural killer (NK) cells. CXCR6þ cells were enriched in thyroid-derived T-cells compared with peripheral CD4þ and CD8þ T-cells in GD. The increase was evident within the Th1 (CD4þ interferon-gþ (IFN-gþ)) and Tc1 (CD8þIFN- gþ) subpopulation and CD8þ granzyme Aþ T-cells (cytotoxic effector type). -

Annex I Summary of Product Characteristics
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT VIRACEPT 250 mg film-coated tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION VIRACEPT 250 mg film-coated tablets contain 292.25 mg of nelfinavir mesylate corresponding to 250 mg of nelfinavir (as free base). For excipients, see 6.1. 3. PHARMACEUTICAL FORM Film-coated tablets 4. CLINICAL PARTICULARS 4.1 Therapeutic indications VIRACEPT is indicated in antiretroviral combination treatment of human immunodeficiency virus (HIV-1) infected adults, adolescents and children of 3 years of age and older. In protease inhibitor experienced patients the choice of nelfinavir should be based on individual viral resistance testing and treatment history. Refer to Section 5.1 Pharmacodynamic properties. 4.2 Posology and method of administration VIRACEPT film-coated tablets are administered orally and should be ingested with food. Patients older than 13 years: the recommended dosage of VIRACEPT film-coated tablets is 1250 mg (five 250 mg tablets) twice a day (BID) or 750 mg (three 250 mg tablets) three times a day (TID) by mouth. The efficacy of the BID regimen has been evaluated versus the TID regimen primarily in patients naïve to protease inhibitors (see section 5.1, pharmacodynamic properties). Patients aged 3 to 13 years: for children, the recommended starting dose is 25 – 30 mg/kg body weight per dose given TID. For children unable to take tablets, VIRACEPT oral powder may be administered (see Summary of Product Characteristics for VIRACEPT oral powder). The recommended dose of VIRACEPT film-coated tablets to be administered TID to children aged 3 to 13 years is as follows: Body Weight Number of Viracept 250mg kg film-coated tablets* 18 to < 23 2 ≥ 23 3 * see Summary of Product Characteristics for VIRACEPT oral powder for patients with less than 18 kg body weight. -

DESCOVY, and Upon Diagnosis of These Highlights Do Not Include All the Information Needed to Use Any Other Sexually Transmitted Infections (Stis)
HIGHLIGHTS OF PRESCRIBING INFORMATION once every 3 months while taking DESCOVY, and upon diagnosis of These highlights do not include all the information needed to use any other sexually transmitted infections (STIs). (2.2) DESCOVY safely and effectively. See full prescribing information • Recommended dosage: for DESCOVY. • Treatment of HIV-1 Infection: One tablet taken once daily with or ® without food in patients with body weight at least 25 kg. (2.3) DESCOVY (emtricitabine and tenofovir alafenamide) tablets, for • HIV-1 PrEP: One tablet taken once daily with or without food in oral use individuals with body weight at least 35 kg. (2.4) Initial U.S. Approval: 2015 • Renal impairment: DESCOVY is not recommended in individuals with WARNING: POST-TREATMENT ACUTE EXACERBATION OF estimated creatinine clearance below 30 mL per minute. (2.5) HEPATITIS B and RISK OF DRUG RESISTANCE WITH USE ----------------------DOSAGE FORMS AND STRENGTHS-------------------- OF DESCOVY FOR HIV-1 PRE-EXPOSURE PROPHYLAXIS Tablets: 200 mg of FTC and 25 mg of TAF (3) (PrEP) IN UNDIAGNOSED EARLY HIV-1 INFECTION See full prescribing information for complete boxed warning. -------------------------------CONTRAINDICATIONS------------------------------ DESCOVY for HIV-1 PrEP is contraindicated in individuals with Severe acute exacerbations of hepatitis B (HBV) have been unknown or positive HIV-1 status. (4) reported in HBV-infected individuals who have discontinued products containing emtricitabine (FTC) and/or tenofovir -----------------------WARNINGS AND PRECAUTIONS----------------------- disoproxil fumarate (TDF), and may occur with • Comprehensive management to reduce the risk of sexually discontinuation of DESCOVY. Hepatic function should be transmitted infections (STIs), including HIV-1, when DESCOVY is monitored closely in these individuals.