Mitochondria Are Sensors for HIV Drugs
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
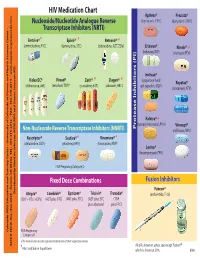
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (Tipranavir, TPV) (Darunavir, DRV) Transcriptase Inhibitors (NRTI)
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (tipranavir, TPV) (darunavir, DRV) Transcriptase Inhibitors (NRTI) Emtriva®* Epivir® * Retrovir® * (emtricitabine, FTC) (lamivudine, 3TC) (zidovudine, AZT, ZDV) Crixivan® Norvir® * (indinavir, IDV) (ritonavir, RTV) Invirase® Videx EC® Viread® Zerit® Ziagen® * * (saquinavir hard Reyataz® (didanosine, ddl) (tenofovir,TDF)* (stavudine, d4T) (abacavir, ABC) gel capsules, SQV) (atazanavir, ATV) Kaletra® * (lopinavir/ritonavir, LPV/r) Viracept® Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) Inhibitors (PI) Protease (nelfinavir, NFV) Rescriptor® Sustiva®* Viramune® * (delavirdine, DLV) (efavirenz, EFV) (nevirapine, NVP) Lexiva® (fosamprenavir, FPV) FDA Pregnancy Category D Fixed Dose Combinations Fusion Inhibitors Fuzeon® Developed by MeriLou Johnson, MeriLou by Developed Johnson, MSW, and Steven MPA MD Atripla® Combivir® Epzicom® Trizivir® Truvada® (enfuvirtide,T-20) (TDF+FTC+EFV) (AZT plus 3TC) (ABC plus 3TC) (AZT plus 3TC (TDF plus abacavir) plus FTC) Colorado AIDS Education and Training Center • University of Colorado at Denver and Health Sciences Center • Center and Health Sciences Denver at of Colorado • University Center Training and AIDS Education Colorado FDA Pregnancy Category D ®The brands listed are the registered trademarks of their respective owners. 4200 E. Ave., Ninth TEL: A089 • Denver, Box 80262 CO 303-315-2512 • FAX: • 303-315-2514 • www.mpaetc.org/colorado.htm All pills shown in actual size except Fuzeon® *Also available in liquid form. which is shown at 50%. 8/06 Medication Schedule Name______________________________ Date___________ Number of Time of day you ar to take TOTAL Name of Medication pills to take this medicine Food Interactions Side Effects number of each time pills each day With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ Discontinued Medications or Formulations Helpful Hints: • Refill prescriptions before you run out. -

Design and Evaluation of 5•²-O-Dicarboxylic And
University of Rhode Island DigitalCommons@URI Open Access Master's Theses 2014 DESIGN AND EVALUATION OF 5′-O-DICARBOXYLIC AND POLYARGININE FATTY ACYL DERIVATIVES OF ANTI-HIV NUCLEOSIDES Bhanu Priya Pemmaraju Venkata University of Rhode Island, [email protected] Follow this and additional works at: https://digitalcommons.uri.edu/theses Recommended Citation Pemmaraju Venkata, Bhanu Priya, "DESIGN AND EVALUATION OF 5′-O-DICARBOXYLIC AND POLYARGININE FATTY ACYL DERIVATIVES OF ANTI-HIV NUCLEOSIDES" (2014). Open Access Master's Theses. Paper 474. https://digitalcommons.uri.edu/theses/474 This Thesis is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Master's Theses by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. DESIGN AND EVALUATION OF 5′-O- DICARBOXYLIC AND POLYARGININE FATTY ACYL DERIVATIVES OF ANTI-HIV NUCLEOSIDES BY BHANU PRIYA, PEMMARAJU VENKATA A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE MASTER’S DEGREE IN BIOMEDICAL AND PHARMACEUTICAL SCIENCES UNIVERSITY OF RHODE ISLAND 2014 MASTER OF SCIENCE THESIS OF BHANU PRIYA, PEMMARAJU VENKATA APPROVED: Thesis Committee: Major Professor Keykavous Parang Roberta King Stephen Kogut Geoffrey D. Bothun Nasser H. Zawia DEAN OF THE GRADUATE SCHOOL UNIVERSITY OF RHODE ISLAND 2014 ABSTRACT 2′,3′-Dideoxynucleoside (ddNs) analogs are the most widely used anti-HIV drugs in the market. Even though these drugs display very potent activities, they have a number of limitations when are used as therapeutic agents. The primary problem associated with ddNs is significant toxicity, such as neuropathy and bone marrow suppression. -

(12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson Et Al
US 20080241135A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson et al. (43) Pub. Date: Oct. 2, 2008 (54) METHODS FOR REDUCINGVIRAL LOAD IN 60/711,528, filed on Aug. 26, 2005, provisional appli HIV-1-INFECTED PATIENTS cation No. 60/715,619, filed on Sep. 9, 2005. (75) Inventors: William C. Olson, Ossining, NY Publication Classification (US); Paul J. Maddon, Scarsdale, NY (US); Daniel C. Pevear, (51) Int. Cl. Medford, MA (US); Robert J. A 6LX 39/395 (2006.01) Israel, Suffern, NY (US); Jose D. A6IP 43/00 (2006.01) Murga, Rosedale, NY (US) (52) U.S. Cl. ..................................................... 424/133.1 Correspondence Address: (57) ABSTRACT COOPER & DUNHAM, LLP 1185 AVENUE OF THE AMERICAS This method provides a method for reducing HIV-1 viral load NEW YORK, NY 10036 in an HIV-1-infected human subject which comprises admin istering to the subject at a predefined interval effective HIV-1 (73) Assignee: Progenics Pharmaceuticals, Inc. viral load-reducing doses of (a) a humanized antibody desig nated PRO 140, or of (b) an anti-CCR5 receptor monoclonal (21) Appl. No.: 11/894,762 antibody. This invention also provides a method for inhibiting in a human Subject the onset or progression of an HIV-1- (22) Filed: Aug. 20, 2007 associated disorder, the inhibition of which is effected by inhibiting fusion of HIV-1 to CCR5"CD4" target cells in the Related U.S. Application Data Subject. This invention also provides a method for treating a subject infected with HIV-1 comprising administering to the (63) Continuation of application No. -

Recent Advances in Antiviral Therapy J Clin Pathol: First Published As 10.1136/Jcp.52.2.89 on 1 February 1999
J Clin Pathol 1999;52:89–94 89 Recent advances in antiviral therapy J Clin Pathol: first published as 10.1136/jcp.52.2.89 on 1 February 1999. Downloaded from Derek Kinchington Abstract indicated that using a combination of drugs In the early 1980s many institutions in might overcome this problem. The only Britain were seriously considering available drugs during the late 1980s were two whether there was a need for specialist other nucleotide reverse transcriptase inhibi- departments of virology. The arrival of tors (NRTI) which also targeted HIV reverse HIV changed that perception and since transcriptase (HIV-RT): 2',3'-dideoxycytidine then virology and antiviral chemotherapy (ddC) and 2',3'-dideoxyinosine (ddI).56 In have become two very active areas of bio- vitro combination studies gave surprising medical research. Cloning and sequencing results: those viruses that became highly resist- have provided tools to identify viral en- ant to ZDV remained sensitive to both ddC zymes and have brought the day of the and ddI.7 Furthermore, neither cross resistance “designer drug” nearer to reality. At the nor interference between the drugs was an other end of the spectrum of drug discov- issue, and subsequent clinical experience ery, huge numbers of compounds for showed that patients benefited when these two screening can now be generated by combi- compounds were used in combination with natorial chemistry. The impetus to find ZDV.8 It was also found by in vitro studies that drugs eVective against HIV has also virus isolated from patients on long term ZDV stimulated research into novel treatments monotherapy had become insensitive to ZDV, for other virus infections including her- but regained sensitivity when these patients pesvirus, respiratory infections, and were switched to ddI monotherapy. -

Guidelines for the Use of Antiretroviral Agents in Adults and Adolescent Living With
Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV Developed by the DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents – A Working Group of the Office of AIDS Research Advisory Council (OARAC) How to Cite the Adult and Adolescent Guidelines: Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services. Available at https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/ AdultandAdolescentGL.pdf. Accessed [insert date] [insert page number, table number, etc. if applicable] It is emphasized that concepts relevant to HIV management evolve rapidly. The Panel has a mechanism to update recommendations on a regular basis, and the most recent information is available on the HIVinfo Web site (http://hivinfo.nih.gov). What’s New in the Guidelines? August 16, 2021 Hepatitis C Virus/HIV Coinfection • Table 18 of this section has been updated to include recommendations regarding concomitant use of fostemsavir or long acting cabotegravir plus rilpivirine with different hepatitis C treatment regimens. June 3, 2021 What to Start • Since the release of the last guidelines, updated data from the Botswana Tsepamo study have shown that the prevalence of neural tube defects (NTD) associated with dolutegravir (DTG) use during conception is much lower than previously reported. Based on these new data, the Panel now recommends that a DTG-based regimen can be prescribed for most people with HIV who are of childbearing potential. Before initiating a DTG-based regimen, clinicians should discuss the risks and benefits of using DTG with persons of childbearing potential, to allow them to make an informed decision. -

Safety and Efficacy of Antiviral Therapy for Prevention of Cytomegalovirus Reactivation in Immunocompetent Critically Ill Patien
1 2 3 PROJECT TITLE 4 Anti-viral Prophylaxis for Prevention of Cytomegalovirus (CMV) Reactivation in Immunocompetent 5 Patients in Critical Care 6 7 STUDY ACRONYM 8 Cytomegalovirus Control in Critical Care - CCCC 9 10 APPLICANTS 11 Dr Nicholas Cowley 12 Specialty Registrar Anaesthesia and Intensive Care Medicine, Intensive Care Research Fellow 13 Queen Elizabeth Hospital Birmingham 14 15 Professor Paul Moss 16 Professor of Haematology 17 Queen Elizabeth Hospital Birmingham 18 19 Professor Julian Bion 20 Professor of Intensive Care Medicine 21 Queen Elizabeth Hospital Birmingham 22 23 Trial Virologist Trial Statistician 24 Dr H Osman Dr P G Nightingale 25 Queen Elizabeth Hospital Birmingham University of Birmingham CCCC CMV Protocol V1.7, 18th September 2013 1 Downloaded From: https://jamanetwork.com/ on 09/23/2021 26 CONTENTS 27 Substantial Amendment Sept 18th 2013 4 28 1 SUMMARY OF TRIAL DESIGN .......................................................................................................... 5 29 2 QEHB ICU Duration of Patient Stay ................................................................................................. 6 30 3 SCHEMA - QEHB PATIENT NUMBERS AVAILABLE FOR RECRUITMENT ........................................... 6 31 4 INTRODUCTION ............................................................................................................................... 7 32 4.1 CMV latent infection is widespread ........................................................................................ 7 33 4.2 CMV Reactivation -

Page: Treatment-Drugs
© National HIV Curriculum PDF created September 27, 2021, 11:42 pm Saquinavir (Invirase) Table of Contents Saquinavir Invirase Summary Drug Summary Key Clinical Trials Resistance Key Drug Interactions Drug Summary Saquinavir, the first protease inhibitor approved by the FDA (in 1995), played a central part in the initial roll out of highly active combination antiretroviral therapy in the mid-1990’s. Three formulations of saquinavir have been approved, including the initial hard-gel capsule, a soft-gel capsule (now discontinued), and, more recently, a tablet. In the early years of combination antiretroviral therapy, unboosted saquinavir was used in combination with two nucleoside reverse transcriptase inhibitors (NRTIs). Subsequently, saquinavir has been used in combination with low-dose ritonavir boosting. Over time, multiple better-tolerated and more convenient antiretroviral agents have become available and have replaced saquinavir. At this time, saquinavir is no longer recommended for use in antiretroviral therapy regimens and patients taking this agent should switch to a currently recommended option. Key Clinical Trials Early trials demonstrated that saquinavir plus two NRTI’s (zidovudine and zalcitabine) led to greater reductions in HIV RNA and increases in CD4 count as compared to dual therapy with zidovudine plus saquinavir or zidovudine plus zalcitabine [ACTG 229]. Subsequently, saquinavir was compared to other protease inhibitors, each given with two NRTIs. One study demonstrated similar antiviral potency between saquinavir boosted with ritonavir and lopinavir-ritonavir, each given with tenofovir DF-emtricitabine [GEMINI], whereas a similar trial demonstrated higher rates of treatment failure and discontinuation in the saquinavir plus ritonavir arm and concluded that lopinavir-ritonavir has superior antiviral efficacy, largely driven by tolerability and patient preference [MaxCmin2]. -

Rational Design and Synthesis of New Nucleoside Analogues Bearing a Cyclohexane Core
Rational Design and Synthesis of New Nucleoside Analogues Bearing a Cyclohexane Core Beatriz Domínguez Pérez Ph.D. Thesis Ph.D. in Chemistry Supervisors: Dr. Ramon Alibés Arqués Dr. Félix Busqué Sánchez Dr. Jean-Didier Márechal Departament de Química Facultat de Ciències 2015 Memòria presentada per aspirar al Grau de Doctor per Beatriz Domínguez Pérez Beatriz Domínguez Pérez Vist i plau, Dr. Ramon Alibés Arqués Dr. Félix Busqué Sánchez Dr. Jean-Didier Maréchal Bellaterra, 13 de maig de 2015 “When you make the finding yourself-even if you’re the last person on Earth to see the light- you’ll never forget it” Carl Sagan A mis padres, a mi hermana A Jonatan Table of contents Abbreviations................................................................................................................................ 1 I. General introduction ............................................................................................. 5 1. Viruses ................................................................................................................................. 7 1.1. Virus replication ............................................................................................................. 8 1.2. Viral diseases in humans ................................................................................................ 9 2. Antiviral drugs ................................................................................................................... 10 2.1. Nucleoside analogues ................................................................................................. -

SELZENTRY (Maraviroc) Tablets, for Oral Use • Hepatotoxicity Accompanied by Severe Rash Or Systemic Allergic Reaction
---------------------------------- CONTRAINDICATIONS --------------------------------- HIGHLIGHTS OF PRESCRIBING INFORMATION • SELZENTRY is contraindicated in patients with severe renal impairment These highlights do not include all the information needed to use or end-stage renal disease (ESRD) (CrCl less than 30 mL per minute) SELZENTRY safely and effectively. See full prescribing information for who are concomitantly taking potent CYP3A inhibitors or inducers. (4) SELZENTRY. -------------------------- WARNINGS AND PRECAUTIONS -------------------------- SELZENTRY (maraviroc) tablets, for oral use • Hepatotoxicity accompanied by severe rash or systemic allergic reaction, SELZENTRY (maraviroc) oral solution including potentially life-threatening events, has been reported. Hepatic Initial U.S. Approval: 2007 laboratory parameters including ALT, AST, and bilirubin should be obtained prior to starting SELZENTRY and at other time points during WARNING: HEPATOTOXICITY See full prescribing information for complete boxed warning. treatment as clinically indicated. If rash or symptoms or signs of hepatitis or allergic reaction develop, hepatic laboratory parameters should be • Hepatotoxicity has been reported which may be preceded by severe monitored and discontinuation of treatment should be considered. When rash or other features of a systemic allergic reaction (e.g., fever, administering SELZENTRY to patients with pre-existing liver eosinophilia, or elevated IgE). (5.1) dysfunction or who are co-infected with hepatitis B and/or C virus, • -

SYMTUZA™ (Darunavir, Cobicistat, Emtricitabine, Tenofovir Alafenamide) Oral
PHARMACY COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 9/20/2018 SECTION: DRUGS LAST REVIEW DATE: 8/19/2021 LAST CRITERIA REVISION DATE: 8/19/2021 ARCHIVE DATE: SYMTUZA™ (darunavir, cobicistat, emtricitabine, tenofovir alafenamide) oral Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Pharmacy Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Pharmacy Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide BCBSAZ complete medical rationale when requesting any exceptions to these guidelines. The section identified as “Description” defines or describes a service, procedure, medical device or drug and is in no way intended as a statement of medical necessity and/or coverage. The section identified as “Criteria” defines criteria to determine whether a service, procedure, medical device or drug is considered medically necessary or experimental or investigational. State or federal mandates, e.g., FEP program, may dictate that any drug, device or biological product approved by the U.S. Food and Drug Administration (FDA) may not be considered experimental or investigational and thus the drug, device or biological product may be assessed only on the basis of medical necessity. Pharmacy Coverage Guidelines are subject to change as new information becomes available. For purposes of this Pharmacy Coverage Guideline, the terms "experimental" and "investigational" are considered to be interchangeable. -

Treatment of AIDS and HIV-Related Conditions: 2001 J Am Board Fam Pract: First Published As on 1 July 2001
Current Report—HIV Treatment of AIDS and HIV-Related Conditions: 2001 J Am Board Fam Pract: first published as on 1 July 2001. Downloaded from Ronald H. Goldschmidt, MD, and Betty J. Dong, PharmD Managing human immunodeficiency virus (HIV) offer the best opportunity to provide comprehen- disease and the acquired immunodeficiency syn- sive care. drome (AIDS) has become more standardized yet This Current Report—HIV updates our annual more complex during the past year. Antiretroviral treatment guidelines.3 These recommendations treatment guidelines now represent a general con- (Table 1) are based on our experience at San Fran- sensus on basic treatment principles and options1 as cisco General Hospital, published guidelines, a re- benefits and risks of therapy have become more view of the medical literature, and experience evident. Clinical manifestations of HIV infection, gained from answering telephone calls to our Na- prognosis, and quality of life are clearly improved tional HIV Telephone Consultation Service for most patients receiving potent antiretroviral (Warmline). Because HIV management changes therapy, yet viral resistance and chronic and short- rapidly, clinicians are advised to refer to the excel- term drug toxicities remain major problems. The lent federal guidelines for the use of antiretroviral markedly decreased incidence of opportunistic in- agents in HIV-infected adults and adolescents1 and fections among treated patients has made unusual for prevention of opportunistic infections,4 which infections less common in daily HIV management. are updated frequently on the Internet (Table 2). The role of resistance assays in HIV care is still These and other federal guidelines are available at being assessed, but they offer great possibilities for http://www.hivatis.org. -

1. Global HIV Infection Drug Market Overview 1.1 Global HIV Incidence Scenario 1.2 Market Overview: Global & Regional 1.3 Clinical Pipeline Overview
August’2014 Global HIV Infection Drug Market & Pipeline Insight Global HIV Infection Drug Market & Pipeline Insight ©KuicK Research Global HIV Infection Drug Market & Pipeline Insight ©KuicK Research All rights reserved. No part of this research study may be reproduced, used, quoted and modified in any form and means without the prior consent of KuicK Research. The use of this research study is only allowed as per the license agreement/purchased from KuicK Research. Global HIV Infection Drug Market & Pipeline Insight ©KuicK Research Page 2 Table of Contents 1. Global HIV Infection Drug Market Overview 1.1 Global HIV Incidence Scenario 1.2 Market Overview: Global & Regional 1.3 Clinical Pipeline Overview 2. Global HIV Infection Drug Market Dynamics 2.1 Favorable Market Drivers 2.2 Market Challenges to be Resolved 2.3 Future Opportunity Outlook 3. FDA Regulatory Framework for Development of HIV Vaccine 3.1 Development of Preventive HIV Vaccines for Use in Paediatric Populations 3.2 Guidance for Submitting HIV Resistance Data 3.3 HIV Resistance Testing in Antiretroviral Drug Development 3.4 HIV-1 Infection: Developing Antiretroviral Drugs for Treatment 4. EMA Regulatory Framework for Development of Medicinal Products for Treatment of HIV Infection 5. Global HIV Infection Drug Clinical Pipeline by Phase, Company & Country 5.1 Phase Unknown 5.2 Research 5.3 Preclinical 5.4 Clinical 5.5 Phase-0 5.6 Phase-I 5.7 Phase-I/II 5.8 Phase-II 5.9 Phase-III 5.10 Preregistration 5.11 Registered Global HIV Infection Drug Market & Pipeline Insight ©KuicK Research Page 3 6.