Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Highlights of Prescribing Information
HIGHLIGHTS OF PRESCRIBING INFORMATION --------------------------WARNINGS AND PRECAUTIONS-------------------- These highlights do not include all the information needed to use • New onset or worsening renal impairment: Can include acute VIREAD safely and effectively. See full prescribing information renal failure and Fanconi syndrome. Assess creatinine clearance for VIREAD. (CrCl) before initiating treatment with VIREAD. Monitor CrCl and ® serum phosphorus in patients at risk. Avoid administering VIREAD (tenofovir disoproxil fumarate) tablets, for oral use VIREAD with concurrent or recent use of nephrotoxic drugs. (5.3) VIREAD® (tenofovir disoproxil fumarate) powder, for oral use • Coadministration with Other Products: Do not use with other Initial U.S. Approval: 2001 tenofovir-containing products (e.g., ATRIPLA, COMPLERA, and TRUVADA). Do not administer in combination with HEPSERA. WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH (5.4) STEATOSIS and POST TREATMENT EXACERBATION OF HEPATITIS • HIV testing: HIV antibody testing should be offered to all HBV- infected patients before initiating therapy with VIREAD. VIREAD See full prescribing information for complete boxed warning. should only be used as part of an appropriate antiretroviral • Lactic acidosis and severe hepatomegaly with steatosis, combination regimen in HIV-infected patients with or without HBV including fatal cases, have been reported with the use of coinfection. (5.5) nucleoside analogs, including VIREAD. (5.1) • Decreases in bone mineral density (BMD): Consider assessment • Severe acute exacerbations of hepatitis have been reported of BMD in patients with a history of pathologic fracture or other in HBV-infected patients who have discontinued anti- risk factors for osteoporosis or bone loss. (5.6) hepatitis B therapy, including VIREAD. Hepatic function • Redistribution/accumulation of body fat: Observed in HIV-infected should be monitored closely in these patients. -
(12) United States Patent (10) Patent No.: US 8,993,581 B2 Perrine Et Al
US00899.3581B2 (12) United States Patent (10) Patent No.: US 8,993,581 B2 Perrine et al. (45) Date of Patent: Mar. 31, 2015 (54) METHODS FOR TREATINGVIRAL (58) Field of Classification Search DSORDERS CPC ... A61K 31/00; A61K 31/166; A61K 31/185: A61K 31/233; A61K 31/522: A61K 38/12: (71) Applicant: Trustees of Boston University, Boston, A61K 38/15: A61K 45/06 MA (US) USPC ........... 514/263.38, 21.1, 557, 565, 575, 617; 424/2011 (72) Inventors: Susan Perrine, Weston, MA (US); Douglas Faller, Weston, MA (US) See application file for complete search history. (73) Assignee: Trustees of Boston University, Boston, (56) References Cited MA (US) U.S. PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this 3,471,513 A 10, 1969 Chinn et al. patent is extended or adjusted under 35 3,904,612 A 9/1975 Nagasawa et al. U.S.C. 154(b) by 0 days. (Continued) (21) Appl. No.: 13/915,092 FOREIGN PATENT DOCUMENTS (22) Filed: Jun. 11, 2013 CA 1209037 A 8, 1986 CA 2303268 A1 4f1995 (65) Prior Publication Data (Continued) US 2014/OO45774 A1 Feb. 13, 2014 OTHER PUBLICATIONS Related U.S. Application Data (63) Continuation of application No. 12/890,042, filed on PCT/US 10/59584 Search Report and Written Opinion mailed Feb. Sep. 24, 2010, now abandoned. 11, 2011. (Continued) (60) Provisional application No. 61/245,529, filed on Sep. 24, 2009, provisional application No. 61/295,663, filed on Jan. 15, 2010. Primary Examiner — Savitha Rao (74) Attorney, Agent, or Firm — Nixon Peabody LLP (51) Int. -

COVID-19) Pandemic on National Antimicrobial Consumption in Jordan
antibiotics Article An Assessment of the Impact of Coronavirus Disease (COVID-19) Pandemic on National Antimicrobial Consumption in Jordan Sayer Al-Azzam 1, Nizar Mahmoud Mhaidat 1, Hayaa A. Banat 2, Mohammad Alfaour 2, Dana Samih Ahmad 2, Arno Muller 3, Adi Al-Nuseirat 4 , Elizabeth A. Lattyak 5, Barbara R. Conway 6,7 and Mamoon A. Aldeyab 6,* 1 Clinical Pharmacy Department, Jordan University of Science and Technology, Irbid 22110, Jordan; [email protected] (S.A.-A.); [email protected] (N.M.M.) 2 Jordan Food and Drug Administration (JFDA), Amman 11181, Jordan; [email protected] (H.A.B.); [email protected] (M.A.); [email protected] (D.S.A.) 3 Antimicrobial Resistance Division, World Health Organization, Avenue Appia 20, 1211 Geneva, Switzerland; [email protected] 4 World Health Organization Regional Office for the Eastern Mediterranean, Cairo 11371, Egypt; [email protected] 5 Scientific Computing Associates Corp., River Forest, IL 60305, USA; [email protected] 6 Department of Pharmacy, School of Applied Sciences, University of Huddersfield, Huddersfield HD1 3DH, UK; [email protected] 7 Institute of Skin Integrity and Infection Prevention, University of Huddersfield, Huddersfield HD1 3DH, UK * Correspondence: [email protected] Citation: Al-Azzam, S.; Mhaidat, N.M.; Banat, H.A.; Alfaour, M.; Abstract: Coronavirus disease 2019 (COVID-19) has overlapping clinical characteristics with bacterial Ahmad, D.S.; Muller, A.; Al-Nuseirat, respiratory tract infection, leading to the prescription of potentially unnecessary antibiotics. This A.; Lattyak, E.A.; Conway, B.R.; study aimed at measuring changes and patterns of national antimicrobial use for one year preceding Aldeyab, M.A. -

Chapter 12 Antimicrobial Therapy Antibiotics
Chapter 12 Antimicrobial Therapy Topics: • Ideal drug - Antimicrobial Therapy - Selective Toxicity • Terminology - Survey of Antimicrobial Drug • Antibiotics - Microbial Drug Resistance - Drug and Host Interaction An ideal antimicrobic: Chemotherapy is the use of any chemical - soluble in body fluids, agent in the treatment of disease. - selectively toxic , - nonallergenic, A chemotherapeutic agent or drug is any - reasonable half life (maintained at a chemical agent used in medical practice. constant therapeutic concentration) An antibiotic agent is usually considered to - unlikely to elicit resistance, be a chemical substance made by a - has a long shelf life, microorganism that can inhibit the growth or - reasonably priced. kill microorganisms. There is no ideal antimicrobic An antimicrobic or antimicrobial agent is Selective Toxicity - Drugs that specifically target a chemical substance similar to an microbial processes, and not the human host’s. antibiotic, but may be synthetic. Antibiotics Spectrum of antibiotics and targets • Naturally occurring antimicrobials – Metabolic products of bacteria and fungi – Reduce competition for nutrients and space • Bacteria – Streptomyces, Bacillus, • Molds – Penicillium, Cephalosporium * * 1 The mechanism of action for different 5 General Mechanisms of Action for antimicrobial drug targets in bacterial cells Antibiotics - Inhibition of Cell Wall Synthesis - Disruption of Cell Membrane Function - Inhibition of Protein Synthesis - Inhibition of Nucleic Acid Synthesis - Anti-metabolic activity Antibiotics -

The Protean Neurologic Manifestations of Varicella-Zoster Virus Infection
REVIEW MARIA A. NAGEL, MD DONALD H. GILDEN, MD Department of Neurology, University of Departments of Neurology and Microbiology, Colorado Health Sciences Center, Denver University of Colorado Health Sciences Center, Denver The protean neurologic manifestations of varicella-zoster virus infection ■ ABSTRACT ARICELLA-ZOSTER VIRUS (VZV) is an V exclusively human, highly neurotropic Multiple neurologic complications may follow the alphaherpesvirus. Primary infection causes reactivation of varicella-zoster virus (VZV), including chickenpox, after which the virus becomes herpes zoster (also known as zoster or shingles), latent in cranial nerve ganglia, dorsal root postherpetic neuralgia, vasculopathy, myelitis, necrotizing ganglia, and autonomic ganglia along the retinitis, and zoster sine herpete (pain without rash). entire neuraxis. Decades later, VZV can reac- These conditions can be difficult to recognize, especially tivate to cause a number of neurologic condi- as several can occur without rash. tions. This article discusses the protean manifes- ■ KEY POINTS tations of VZV reactivation and their diagno- sis and treatment. The most common sequela of herpes zoster is postherpetic neuralgia, which can persist for months and ■ VZV REACTIVATION sometimes years after the rash resolves. HAS MANY MANIFESTATIONS VZV vasculopathy can present as transient ischemic VZV reactivation can produce a number of neurologic complications: attacks, ischemic or hemorrhagic stroke, or aneurysm. •Herpes zoster (shingles), manifesting as pain and rash in up to three dermatomes Vasculopathy and neurologic complications of VZV are • Postherpetic neuralgia, the most common best diagnosed by detecting VZV DNA in bodily fluids or sequela of zoster, is pain that persists for tissues or anti-VZV immunoglobulin G in the months and sometimes years after the rash cerebrospinal fluid. -

“Amenalief® Tab. 200Mg” in Japan
News Release July 3, 2017 Maruho Co., Ltd. Maruho receives manufacturing and marketing approval for Anti-Herpes Virus Agent “Amenalief® Tab. 200mg” in Japan Osaka (Japan), July 3, 2017 – Maruho Co., Ltd (“Maruho”, Head Office: Osaka, Japan, President and CEO: Koichi Takagi) announces that today it has received manufacturing and marketing approval from the Japanese Ministry of Health, Labor and Welfare (MHLW), for anti-herpes virus agent “Amenalief® Tab. 200mg” (INN: amenamevir) (hereinafter referred to as “the product”) for the treatment of herpes zoster (shingles) in Japan. The product is an anti-herpes virus agent with a novel mechanism of action created by Astellas Pharma Inc. (“Astellas”; Head Office: Tokyo, President and CEO: Yoshihiko Hatanaka). The product has been observed to inhibit the proliferation of the varicella-zoster virus (hereinafter VZV) by inhibiting the activity of the helicase-primase complex, which is essential for viral DNA replication. In August 2012, Maruho and Astellas agreed on a license agreement for the development and commercialization of the product in Japan, and Maruho has been progressing its development. Herpes zoster (shingles) is a disease caused by reactivation of the chickenpox VZV in latently-infected nerve ganglia. The main treatment for shingles is anti-herpes virus agents. The product is proven to be effective against VZV when administered once a day after meals. Also, since most of the product is excreted in feces, it is not necessary to adjust the dosage and administration according to creatinine clearance, an indicator of kidney function. Maruho hopes the product will contribute to the expansion of treatment options and improvement in adherence for the treatment of shingles in Japan. -
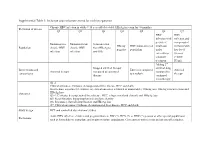
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

Which Drugs Are Most Effective for Recurrent Herpes Labialis?
Evidence-based answers from the clinical inquiries Family Physicians Inquiries Network Eiko Tubridy, MD; Gary Kelsberg, MD Valley Family Residency Which drugs are most Program, Renton, Wash Leilani St Anna, MLIS, effective for recurrent AHIP University of Washington Health Sciences Libraries, herpes labialis? Seattle AssistanT EDITOR EvidEncE-basEd answEr Jon O. neher, MD Valley Family Residency daily oral acyclovir or vala- ing to the agent used: valacyclovir reduces Program, Renton, Wash A cyclovir may help prevent her- both healing time and duration of pain, pes simplex labialis (HSL) recurrences famciclovir reduces both in one dosage (strength of recommendation [SOR]: B, form but not another, and acyclovir reduces meta-analysis of randomized controlled only pain duration (SOR: B, single RCTs). trials [RCTs] with heterogeneous results). Several topical medications (acyclovir, No trials compare oral or topical treat- penciclovir, docosanol) modestly decrease ments for HSL outbreaks against each oth- healing time and pain duration—typically er. Oral antivirals modestly reduce healing by less than a day—and require multiple time and duration of pain, varying accord- doses per day (SOR: B, multiple RCTs). Evidence summary The authors of the meta-analysis noted A systematic review and meta-analysis of the that although 9 studies favored the use of an effectiveness of oral and topical nucleoside antiviral drug, only 4 showed statistically sig- antiviral agents to prevent recurrent HSL in nificant differences when compared with pla- immunocompetent people found 11 RCTs cebo, and none of them had a low risk of bias. with a total of 1250 patients that compared They concluded that the review supported us- an active drug against placebo.1 The medi- ing oral acyclovir and valacyclovir to prevent cations were topical 5% acyclovir, topical 1% recurrent HSL.1 penciclovir, and oral acyclovir, valacyclovir, or famciclovir in various doses. -

CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene and Pathophysiological Conditions
Review CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene and Pathophysiological Conditions Rossana Roncato 1,*,†, Jacopo Angelini 2,†, Arianna Pani 2,3,†, Erika Cecchin 1, Andrea Sartore-Bianchi 2,4, Salvatore Siena 2,4, Elena De Mattia 1, Francesco Scaglione 2,3,‡ and Giuseppe Toffoli 1,‡ 1 1 Experimental and Clinical Pharmacology Unit, Centro di Riferimento Oncologico (CRO), IRCCS, 33081 Aviano, Italy; [email protected] (E.C.); [email protected] (E.D.M.); [email protected] (G.T.) 2 Department of Oncology and Hemato-Oncology, Università degli Studi di Milano, 20122 Milan, Italy; [email protected] (J.A.); [email protected] (A.P.); [email protected] (A.S-B.); [email protected] (S.S.); [email protected] (F.S.) 3 Clinical Pharmacology Unit, ASST Grande Ospedale Metropolitano Niguarda, Piazza dell'Ospedale Maggiore 3, 20162 Milan, Italy 4 Department of Hematology and Oncology, Niguarda Cancer Center, Grande Ospedale Metropolitano Niguarda, 20162 Milan, Italy * Correspondence: [email protected]; Tel.:+390434659130 † These authors contributed equally. ‡ These authors share senior authorship. Int. J. Mol. Sci. 2020, 21, x; doi: www.mdpi.com/journal/ijms Int. J. Mol. Sci. 2020, 21, x 2 of 8 Table S1. Co-administered agents categorized according to their potential risk for Drug-Drug interaction (DDI) in combination with CDK4/6 inhibitors (CDKis). Colors suggest the risk of DDI with CDKis: green, low risk DDI; orange, moderate risk DDI; red, high risk DDI. ADME, absorption, distribution, metabolism, and excretion; GI, Gastrointestinal; TdP, Torsades de Pointes; NTI, narrow therapeutic index. * Cardiological toxicity should be considered especially for ribociclib due to the QT prolongation. -

Aicuris Granted Fast Track Designation by U.S. FDA for Oral Pritelivir for Treatment of HSV Infections in Immunocompromised Adults
AiCuris Granted Fast Track Designation by U.S. FDA for Oral Pritelivir for Treatment of HSV Infections in Immunocompromised Adults Wuppertal, August 01, 2017 - AiCuris Anti-infective Cures GmbH, a leading company in the discovery and development of drugs against infectious diseases, today announced that the Company has been granted Fast Track designation by the U.S. Food and Drug Administration (FDA) for oral pritelivir, AiCuris’ lead candidate for the treatment of acyclovir-resistant mucocutaneous herpes simplex virus (HSV) infections in immunocompromised adults. Fast track is a process designed to facilitate the development, expedite the review and accelerate the approval process of drugs to treat serious conditions and fill an unmet medical need, with the purpose of getting important new drugs to patients sooner. Oral pritelivir, a small molecule helicase-primase inhibitor with a novel mode of action, is currently in a clinical phase 2 study, called PRIOH-1, in the U.S. to evaluate the product candidate’s efficacy and safety compared to i.v. foscarnet, a virostatic agent which is used mainly for the treatment of herpes viruses resistant to other antiviral drugs. In a prior phase 2 study, oral pritelivir showed to significantly improve the suppression of viral shedding compared to the current standard of care for genital HSV-2 infections, the nucleoside analog valacyclovir. The results of this study were published in the Journal of the American Medical Association (JAMA) earlier this year. “The decision by the FDA to grant fast track designation to oral pritelivir underscores that our product might fill the major need for innovative, more efficacious therapies for immunocompromised patients with HSV infections that have become resistant to standard treatments,” said Dr. -

Novel Therapeutics for Epstein–Barr Virus
molecules Review Novel Therapeutics for Epstein–Barr Virus Graciela Andrei *, Erika Trompet and Robert Snoeck Laboratory of Virology and Chemotherapy, Department of Microbiology and Immunology, Rega Institute for Medical Research, KU Leuven, 3000 Leuven, Belgium; [email protected] (E.T.); [email protected] (R.S.) * Correspondence: [email protected]; Tel.: +32-16-321-915 Academic Editor: Stefano Aquaro Received: 15 February 2019; Accepted: 4 March 2019; Published: 12 March 2019 Abstract: Epstein–Barr virus (EBV) is a human γ-herpesvirus that infects up to 95% of the adult population. Primary EBV infection usually occurs during childhood and is generally asymptomatic, though the virus can cause infectious mononucleosis in 35–50% of the cases when infection occurs later in life. EBV infects mainly B-cells and epithelial cells, establishing latency in resting memory B-cells and possibly also in epithelial cells. EBV is recognized as an oncogenic virus but in immunocompetent hosts, EBV reactivation is controlled by the immune response preventing transformation in vivo. Under immunosuppression, regardless of the cause, the immune system can lose control of EBV replication, which may result in the appearance of neoplasms. The primary malignancies related to EBV are B-cell lymphomas and nasopharyngeal carcinoma, which reflects the primary cell targets of viral infection in vivo. Although a number of antivirals were proven to inhibit EBV replication in vitro, they had limited success in the clinic and to date no antiviral drug has been approved for the treatment of EBV infections. We review here the antiviral drugs that have been evaluated in the clinic to treat EBV infections and discuss novel molecules with anti-EBV activity under investigation as well as new strategies to treat EBV-related diseases. -

Multipurpose Tenofovir Disoproxil Fumarate Electrospun Fibers for the Prevention of HIV-1 and HSV-2 Infections
University of Louisville ThinkIR: The University of Louisville's Institutional Repository Electronic Theses and Dissertations 8-2016 Multipurpose tenofovir disoproxil fumarate electrospun fibers for the prevention of HIV-1 and HSV-2 infections. Kevin Tyo Follow this and additional works at: https://ir.library.louisville.edu/etd Part of the Other Chemicals and Drugs Commons, Polymer Chemistry Commons, Preventive Medicine Commons, and the Virus Diseases Commons Recommended Citation Tyo, Kevin, "Multipurpose tenofovir disoproxil fumarate electrospun fibers for the prevention of HIV-1 and HSV-2 infections." (2016). Electronic Theses and Dissertations. Paper 2486. https://doi.org/10.18297/etd/2486 This Master's Thesis is brought to you for free and open access by ThinkIR: The University of Louisville's Institutional Repository. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of ThinkIR: The University of Louisville's Institutional Repository. This title appears here courtesy of the author, who has retained all other copyrights. For more information, please contact [email protected]. MULTIPURPOSE TENOFOVIR DISOPROXIL FUMARATE ELECTROSPUN FIBERS FOR THE PREVENTION OF HIV-1 AND HSV-2 INFECTIONS By Kevin Tyo B.S. Virginia Tech, 2010 A Thesis Submitted to the Faculty of the School of Medicine of the University of Louisville In Partial Fulfillment of the Requirement for the Degree of Master of Science In Pharmacology and Toxicology Department of Pharmacology and Toxicology School of Medicine University of Louisville Louisville, KY August, 2016 MULTIPURPOSE TENOFOVIR DISOPROXIL FUMARATE ELECTROSPUN FIBERS FOR THE PREVENTION OF HIV-1 AND HSV-2 INFECTIONS By Kevin Tyo B.S.