Design and Evaluation of 5•²-O-Dicarboxylic And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biochemical Engineering: Ta Da! a Way to Add Fluorine to Natural Products
RESEARCH HIGHLIGHTS IN BRIEF BIOCHEMICAL ENGINEERING Ta da! A way to add fluorine to natural products Adding fluorine to small molecules can improve their properties, such as preventing enzymatic breakdown and increasing membrane permeation, but it is hard to add fluorine to natural products. Walker et al. engineered polyketide synthase enzymes, which are normally involved in acetate-based biosynthesis, to incorporate fluoroacetate: the primary product of the only biological fluorination route. In Escherichia coli cells, this process was used as a building block to introduce fluorine substituents in a site-selective manner into polyketide-based natural product scaffolds. ORIGINAL RESEARCH PAPER Walker, M. C. et al. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science 341, 1089–1094 (2013) INFECTIOUS DISEASE Combating visceral leishmaniasis Visceral leishmaniasis is often fatal, yet current drugs are very toxic and incur resistance. Because Leishmania spp. require exogenous haem for growth, Guha et al. investigated whether haem acquisition could be a potential target. They found that the haemoglobin receptor (HbR) was conserved across several strains of Leishmania, and a HbR-specific antibody was detected in patients with visceral leishmaniasis. Immunization of mice and hamsters with HbR DNA completely protected against virulent Leishmania donovani infection and stimulated the production of protective cytokines as well as CD4+ and CD8+ T cells, which suggests that HbR is a target for vaccine development. ORIGINAL RESEARCH PAPER Guha, R. et al. Vaccination with Leishmania hemoglobin receptor-encoding DNA protects against visceral leishmanias. Sci. Transl. Med. 5, 202ra121 (2013) G PROTEIN-COUPLED RECEPTORS Crystal structures aid ligand mechanistics Two new papers on G protein-coupled receptor (GPCR) structure shed new light on how different ligands interact with their cognate GPCRs. -

Recent Advances in Antiviral Therapy J Clin Pathol: First Published As 10.1136/Jcp.52.2.89 on 1 February 1999
J Clin Pathol 1999;52:89–94 89 Recent advances in antiviral therapy J Clin Pathol: first published as 10.1136/jcp.52.2.89 on 1 February 1999. Downloaded from Derek Kinchington Abstract indicated that using a combination of drugs In the early 1980s many institutions in might overcome this problem. The only Britain were seriously considering available drugs during the late 1980s were two whether there was a need for specialist other nucleotide reverse transcriptase inhibi- departments of virology. The arrival of tors (NRTI) which also targeted HIV reverse HIV changed that perception and since transcriptase (HIV-RT): 2',3'-dideoxycytidine then virology and antiviral chemotherapy (ddC) and 2',3'-dideoxyinosine (ddI).56 In have become two very active areas of bio- vitro combination studies gave surprising medical research. Cloning and sequencing results: those viruses that became highly resist- have provided tools to identify viral en- ant to ZDV remained sensitive to both ddC zymes and have brought the day of the and ddI.7 Furthermore, neither cross resistance “designer drug” nearer to reality. At the nor interference between the drugs was an other end of the spectrum of drug discov- issue, and subsequent clinical experience ery, huge numbers of compounds for showed that patients benefited when these two screening can now be generated by combi- compounds were used in combination with natorial chemistry. The impetus to find ZDV.8 It was also found by in vitro studies that drugs eVective against HIV has also virus isolated from patients on long term ZDV stimulated research into novel treatments monotherapy had become insensitive to ZDV, for other virus infections including her- but regained sensitivity when these patients pesvirus, respiratory infections, and were switched to ddI monotherapy. -

Guidelines for the Use of Antiretroviral Agents in Adults and Adolescent Living With
Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV Developed by the DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents – A Working Group of the Office of AIDS Research Advisory Council (OARAC) How to Cite the Adult and Adolescent Guidelines: Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services. Available at https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/ AdultandAdolescentGL.pdf. Accessed [insert date] [insert page number, table number, etc. if applicable] It is emphasized that concepts relevant to HIV management evolve rapidly. The Panel has a mechanism to update recommendations on a regular basis, and the most recent information is available on the HIVinfo Web site (http://hivinfo.nih.gov). What’s New in the Guidelines? August 16, 2021 Hepatitis C Virus/HIV Coinfection • Table 18 of this section has been updated to include recommendations regarding concomitant use of fostemsavir or long acting cabotegravir plus rilpivirine with different hepatitis C treatment regimens. June 3, 2021 What to Start • Since the release of the last guidelines, updated data from the Botswana Tsepamo study have shown that the prevalence of neural tube defects (NTD) associated with dolutegravir (DTG) use during conception is much lower than previously reported. Based on these new data, the Panel now recommends that a DTG-based regimen can be prescribed for most people with HIV who are of childbearing potential. Before initiating a DTG-based regimen, clinicians should discuss the risks and benefits of using DTG with persons of childbearing potential, to allow them to make an informed decision. -

Safety and Efficacy of Antiviral Therapy for Prevention of Cytomegalovirus Reactivation in Immunocompetent Critically Ill Patien
1 2 3 PROJECT TITLE 4 Anti-viral Prophylaxis for Prevention of Cytomegalovirus (CMV) Reactivation in Immunocompetent 5 Patients in Critical Care 6 7 STUDY ACRONYM 8 Cytomegalovirus Control in Critical Care - CCCC 9 10 APPLICANTS 11 Dr Nicholas Cowley 12 Specialty Registrar Anaesthesia and Intensive Care Medicine, Intensive Care Research Fellow 13 Queen Elizabeth Hospital Birmingham 14 15 Professor Paul Moss 16 Professor of Haematology 17 Queen Elizabeth Hospital Birmingham 18 19 Professor Julian Bion 20 Professor of Intensive Care Medicine 21 Queen Elizabeth Hospital Birmingham 22 23 Trial Virologist Trial Statistician 24 Dr H Osman Dr P G Nightingale 25 Queen Elizabeth Hospital Birmingham University of Birmingham CCCC CMV Protocol V1.7, 18th September 2013 1 Downloaded From: https://jamanetwork.com/ on 09/23/2021 26 CONTENTS 27 Substantial Amendment Sept 18th 2013 4 28 1 SUMMARY OF TRIAL DESIGN .......................................................................................................... 5 29 2 QEHB ICU Duration of Patient Stay ................................................................................................. 6 30 3 SCHEMA - QEHB PATIENT NUMBERS AVAILABLE FOR RECRUITMENT ........................................... 6 31 4 INTRODUCTION ............................................................................................................................... 7 32 4.1 CMV latent infection is widespread ........................................................................................ 7 33 4.2 CMV Reactivation -
14-258 Phrma HIV/AIDS2014 0819.Indd
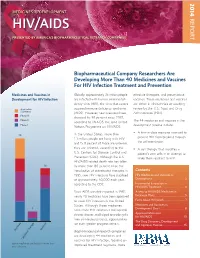
2014 MEDICINES IN DEVELOPMENT REPORT HIV/AIDS PRESENTED BY AMERICA’S BIOPHARMACEUTICAL RESEARCH COMPANIES Biopharmaceutical Company Researchers Are Developing More Than 40 Medicines and Vaccines For HIV Infection Treatment and Prevention Medicines and Vaccines in Globally, approximately 35 million people effective therapies, and preventative Development for HIV Infection are infected with human immunodefi - vaccines. These medicines and vaccines ciency virus (HIV), the virus that causes are either in clinical trials or awaiting Application acquired immune defi ciency syndrome review by the U.S. Food and Drug Submitted (AIDS). However, new infections have Administration (FDA). Phase III dropped by 38 percent since 2001, Phase II The 44 medicines and vaccines in the according to UNAIDS, the Joint United Phase I development pipeline include: Nations Programme on HIV/AIDS. • A fi rst-in-class medicine intended to In the United States, more than 25 prevent HIV from breaking through 1.1 million people are living with HIV the cell membrane. and 15.8 percent of those are unaware they are infected, according to the • A cell therapy that modifi es a U.S. Centers for Disease Control and patient’s own cells in an attempt to Prevention (CDC). Although the U.S. make them resistant to HIV. HIV/AIDS-related death rate has fallen 16 by more than 80 percent since the introduction of antiretroviral therapies in Contents 1995, new HIV infections have stabilized HIV Medicines and Vaccines in at approximately 50,000 each year, Development ......................................2 according to the CDC. Incremental Innovation in HIV/AIDS Treatment .......................... 4 Since AIDS was fi rst reported in 1981, Access to HIV/AIDS Medicines in nearly 40 medicines have been approved Exchange Plans ...................................5 to treat HIV infection in the United Facts About HIV/AIDS ........................7 States. -

Management of Hiv Infection
MANAGEMENT OF HIV INFECTION Federal Bureau of Prisons Clinical Guidance September 2018 Clinical guidance is made available to the public for informational purposes only. The Federal Bureau of Prisons (BOP) does not warrant this guidance for any other purpose, and assumes no responsibility for any injury or damage resulting from the reliance thereof. Proper medical practice necessitates that all cases are evaluated on an individual basis and that treatment decisions are patient-specific. Referenced Program Statement versions within this document are for informational purposes only. Please refer to the most current version of the referenced Program Statement. Consult the BOP Health Management Resources Web page to determine the date of the most recent update to this document: http://www.bop.gov/resources/health_care_mngmt.jsp Federal Bureau of Prisons Management of HIV Infection Clinical Guidance September 2018 WHAT’S NEW IN THIS DOCUMENT? This document updates the December 2017 version of the BOP Clinical Guidance on the Management of HIV Infection. Treatment information throughout this document was updated to be in line with the Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents issued by the Department of Health and Human Services (DHHS) in 2018. NOTEWORTHY CHANGES INCLUDE REVISIONS IN THE FOLLOWING AREAS: • INDICATIONS FOR HIV TESTING: An opt out strategy of voluntary testing for HIV infection is recommended for all inmates, regardless of sentencing status, including new intakes and those already established in the population who have not declined testing. • MENINGOCOCCAL VACCINE: Adults with HIV infection who have not been previously vaccinated should receive a 2-dose primary series of MenACWY (MCV4), available as Menactra® or Menveo®, at least 2 months apart, and then revaccinated every 5 years. -

Rational Design and Synthesis of New Nucleoside Analogues Bearing a Cyclohexane Core
Rational Design and Synthesis of New Nucleoside Analogues Bearing a Cyclohexane Core Beatriz Domínguez Pérez Ph.D. Thesis Ph.D. in Chemistry Supervisors: Dr. Ramon Alibés Arqués Dr. Félix Busqué Sánchez Dr. Jean-Didier Márechal Departament de Química Facultat de Ciències 2015 Memòria presentada per aspirar al Grau de Doctor per Beatriz Domínguez Pérez Beatriz Domínguez Pérez Vist i plau, Dr. Ramon Alibés Arqués Dr. Félix Busqué Sánchez Dr. Jean-Didier Maréchal Bellaterra, 13 de maig de 2015 “When you make the finding yourself-even if you’re the last person on Earth to see the light- you’ll never forget it” Carl Sagan A mis padres, a mi hermana A Jonatan Table of contents Abbreviations................................................................................................................................ 1 I. General introduction ............................................................................................. 5 1. Viruses ................................................................................................................................. 7 1.1. Virus replication ............................................................................................................. 8 1.2. Viral diseases in humans ................................................................................................ 9 2. Antiviral drugs ................................................................................................................... 10 2.1. Nucleoside analogues ................................................................................................. -
![Emtricitabine/Rilpivirine/Tenofovir Alafenamide Fixed-Dose Combination (Ftc/Rpv/Taf [F/R/Taf] Fdc)](https://docslib.b-cdn.net/cover/7965/emtricitabine-rilpivirine-tenofovir-alafenamide-fixed-dose-combination-ftc-rpv-taf-f-r-taf-fdc-1547965.webp)
Emtricitabine/Rilpivirine/Tenofovir Alafenamide Fixed-Dose Combination (Ftc/Rpv/Taf [F/R/Taf] Fdc)
SECTION 2.6.—NONCLINICAL SUMMARY SECTION 2.6.1—INTRODUCTION EMTRICITABINE/RILPIVIRINE/TENOFOVIR ALAFENAMIDE FIXED-DOSE COMBINATION (FTC/RPV/TAF [F/R/TAF] FDC) Gilead Sciences 11 May 2015 CONFIDENTIAL AND PROPRIETARY INFORMATION FTC/RPV/Tenofovir alafenamide 2.6.1 Introduction Final TABLE OF CONTENTS SECTION 2.6.1—INTRODUCTION ...........................................................................................................................1 TABLE OF CONTENTS ..............................................................................................................................................2 GLOSSARY OF ABBREVIATIONS AND DEFINITION OF TERMS......................................................................3 NOTE TO REVIEWER.................................................................................................................................................4 1. NONCLINICAL SUMMARY..............................................................................................................................6 1.1. Introduction..............................................................................................................................................6 1.1.1. Emtricitabine ..........................................................................................................................6 1.1.2. Rilpivirine ..............................................................................................................................7 1.1.3. Tenofovir Alafenamide ..........................................................................................................7 -

1. Global HIV Infection Drug Market Overview 1.1 Global HIV Incidence Scenario 1.2 Market Overview: Global & Regional 1.3 Clinical Pipeline Overview
August’2014 Global HIV Infection Drug Market & Pipeline Insight Global HIV Infection Drug Market & Pipeline Insight ©KuicK Research Global HIV Infection Drug Market & Pipeline Insight ©KuicK Research All rights reserved. No part of this research study may be reproduced, used, quoted and modified in any form and means without the prior consent of KuicK Research. The use of this research study is only allowed as per the license agreement/purchased from KuicK Research. Global HIV Infection Drug Market & Pipeline Insight ©KuicK Research Page 2 Table of Contents 1. Global HIV Infection Drug Market Overview 1.1 Global HIV Incidence Scenario 1.2 Market Overview: Global & Regional 1.3 Clinical Pipeline Overview 2. Global HIV Infection Drug Market Dynamics 2.1 Favorable Market Drivers 2.2 Market Challenges to be Resolved 2.3 Future Opportunity Outlook 3. FDA Regulatory Framework for Development of HIV Vaccine 3.1 Development of Preventive HIV Vaccines for Use in Paediatric Populations 3.2 Guidance for Submitting HIV Resistance Data 3.3 HIV Resistance Testing in Antiretroviral Drug Development 3.4 HIV-1 Infection: Developing Antiretroviral Drugs for Treatment 4. EMA Regulatory Framework for Development of Medicinal Products for Treatment of HIV Infection 5. Global HIV Infection Drug Clinical Pipeline by Phase, Company & Country 5.1 Phase Unknown 5.2 Research 5.3 Preclinical 5.4 Clinical 5.5 Phase-0 5.6 Phase-I 5.7 Phase-I/II 5.8 Phase-II 5.9 Phase-III 5.10 Preregistration 5.11 Registered Global HIV Infection Drug Market & Pipeline Insight ©KuicK Research Page 3 6. -

The Entry of Entry Inhibitors: a Fusion of Science and Medicine
The entry of entry inhibitors: A fusion of science and medicine John P. Moore* and Robert W. Doms†‡ *Department of Microbiology and Immunology, Weill Medical College of Cornell University, 1300 York Avenue, W-805, New York, NY 10021; and †Department of Pathology and Laboratory Medicine, University of Pennsylvania, 806 Abramson, 34th and Civic Center Boulevard, Philadelphia, PA 19104 For HIV-1 to enter a cell, its envelope protein (Env) must sequentially engage CD4 and a chemokine coreceptor, triggering conforma- tional changes in Env that ultimately lead to fusion between the viral and host cell membranes. Each step of the virus entry path- way is a potential target for novel antiviral agents termed entry inhibitors. A growing number of entry inhibitors are under clinical development, with one having already been licensed by the Food and Drug Administration. With the emergence of virus strains that are largely resistant to existing reverse transcriptase and protease inhibitors, the development of entry inhibitors comes at an oppor- tune time. Nonetheless, because all entry inhibitors target in some manner the highly variable Env protein of HIV-1, there are likely to be challenges in their efficient application that are unique to this class of drugs. Env density, receptor expression levels, and dif- ferences in affinity and receptor presentation are all factors that could influence the clinical response to this promising class of new antiviral agents. new class of anti-HIV-1 drugs ing of the receptor-induced conforma- then inserts into it (8). These processes has been developed: com- tional changes in the viral envelope seem to be mediated in part by the for- pounds known variously as fu- (Env) protein that lead to virus-cell fu- mation of a triple-stranded coiled coil sion or entry inhibitors (1, 2). -

EUROPEAN PHARMACOPOEIA 10.0 Index 1. General Notices
EUROPEAN PHARMACOPOEIA 10.0 Index 1. General notices......................................................................... 3 2.2.66. Detection and measurement of radioactivity........... 119 2.1. Apparatus ............................................................................. 15 2.2.7. Optical rotation................................................................ 26 2.1.1. Droppers ........................................................................... 15 2.2.8. Viscosity ............................................................................ 27 2.1.2. Comparative table of porosity of sintered-glass filters.. 15 2.2.9. Capillary viscometer method ......................................... 27 2.1.3. Ultraviolet ray lamps for analytical purposes............... 15 2.3. Identification...................................................................... 129 2.1.4. Sieves ................................................................................. 16 2.3.1. Identification reactions of ions and functional 2.1.5. Tubes for comparative tests ............................................ 17 groups ...................................................................................... 129 2.1.6. Gas detector tubes............................................................ 17 2.3.2. Identification of fatty oils by thin-layer 2.2. Physical and physico-chemical methods.......................... 21 chromatography...................................................................... 132 2.2.1. Clarity and degree of opalescence of -

Review Nucleoside Analogue-Sparing Strategy for the Treatment of Chronic HIV Infection: Potential Interest and Clinical Experience Véronique Joly* and Patrick Yeni
Antiviral Therapy 10:29–40 Review Nucleoside analogue-sparing strategy for the treatment of chronic HIV infection: potential interest and clinical experience Véronique Joly* and Patrick Yeni Maladies Infectieuses, Hôpital Bichat Claude Bernard, Paris, France *Corresponding author: Tel: +33 1 4025 7807; Fax: +33 1 4025 6775; E-mail: [email protected] Nucleoside analogue-sparing antiretroviral combinations therapeutic drug monitoring service to confirm that appro- may be interesting as first-line therapies as they spare a priate drug exposures are achieved is useful when using complete class of drugs that will remain fully active for such regimens. Some negative kinetic interactions may lead later use and prevent the risk of mitochondrial toxicity to complicated combinations with a high pill burden that related to exposure to nucleoside reverse transcriptase reduces their applicability. Gastrointestinal toxicity often inhibitors (NRTIs). This strategy is also used in patients remains a limiting factor in the use of boosted double-PI failing NRTIs with cross-resistance to compounds in this combinations. Non-comparative studies have allowed class. Different combinations of antiretroviral drugs are selection of NRTI-sparing options that now need to be theoretically available. Non-nucleoside reverse transcrip- compared with the current standard of care in comparative tase inhibitors (NNRTIs) associated with protease inhibitor clinical trials before being considered as valuable options. (PI) and boosted double-PI combinations have been Other NRTI-sparing therapeutic strategies are emerging: studied through small, non-comparative clinical studies PI monotherapy with lopinavir/ritonavir has been evalu- and preliminary results suggest that they are efficient and ated in a small group of naive patients and appears often well-tolerated.