The Entry of Entry Inhibitors: a Fusion of Science and Medicine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Design and Evaluation of 5•²-O-Dicarboxylic And
University of Rhode Island DigitalCommons@URI Open Access Master's Theses 2014 DESIGN AND EVALUATION OF 5′-O-DICARBOXYLIC AND POLYARGININE FATTY ACYL DERIVATIVES OF ANTI-HIV NUCLEOSIDES Bhanu Priya Pemmaraju Venkata University of Rhode Island, [email protected] Follow this and additional works at: https://digitalcommons.uri.edu/theses Recommended Citation Pemmaraju Venkata, Bhanu Priya, "DESIGN AND EVALUATION OF 5′-O-DICARBOXYLIC AND POLYARGININE FATTY ACYL DERIVATIVES OF ANTI-HIV NUCLEOSIDES" (2014). Open Access Master's Theses. Paper 474. https://digitalcommons.uri.edu/theses/474 This Thesis is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Master's Theses by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. DESIGN AND EVALUATION OF 5′-O- DICARBOXYLIC AND POLYARGININE FATTY ACYL DERIVATIVES OF ANTI-HIV NUCLEOSIDES BY BHANU PRIYA, PEMMARAJU VENKATA A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE MASTER’S DEGREE IN BIOMEDICAL AND PHARMACEUTICAL SCIENCES UNIVERSITY OF RHODE ISLAND 2014 MASTER OF SCIENCE THESIS OF BHANU PRIYA, PEMMARAJU VENKATA APPROVED: Thesis Committee: Major Professor Keykavous Parang Roberta King Stephen Kogut Geoffrey D. Bothun Nasser H. Zawia DEAN OF THE GRADUATE SCHOOL UNIVERSITY OF RHODE ISLAND 2014 ABSTRACT 2′,3′-Dideoxynucleoside (ddNs) analogs are the most widely used anti-HIV drugs in the market. Even though these drugs display very potent activities, they have a number of limitations when are used as therapeutic agents. The primary problem associated with ddNs is significant toxicity, such as neuropathy and bone marrow suppression. -

Biochemical Engineering: Ta Da! a Way to Add Fluorine to Natural Products
RESEARCH HIGHLIGHTS IN BRIEF BIOCHEMICAL ENGINEERING Ta da! A way to add fluorine to natural products Adding fluorine to small molecules can improve their properties, such as preventing enzymatic breakdown and increasing membrane permeation, but it is hard to add fluorine to natural products. Walker et al. engineered polyketide synthase enzymes, which are normally involved in acetate-based biosynthesis, to incorporate fluoroacetate: the primary product of the only biological fluorination route. In Escherichia coli cells, this process was used as a building block to introduce fluorine substituents in a site-selective manner into polyketide-based natural product scaffolds. ORIGINAL RESEARCH PAPER Walker, M. C. et al. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science 341, 1089–1094 (2013) INFECTIOUS DISEASE Combating visceral leishmaniasis Visceral leishmaniasis is often fatal, yet current drugs are very toxic and incur resistance. Because Leishmania spp. require exogenous haem for growth, Guha et al. investigated whether haem acquisition could be a potential target. They found that the haemoglobin receptor (HbR) was conserved across several strains of Leishmania, and a HbR-specific antibody was detected in patients with visceral leishmaniasis. Immunization of mice and hamsters with HbR DNA completely protected against virulent Leishmania donovani infection and stimulated the production of protective cytokines as well as CD4+ and CD8+ T cells, which suggests that HbR is a target for vaccine development. ORIGINAL RESEARCH PAPER Guha, R. et al. Vaccination with Leishmania hemoglobin receptor-encoding DNA protects against visceral leishmanias. Sci. Transl. Med. 5, 202ra121 (2013) G PROTEIN-COUPLED RECEPTORS Crystal structures aid ligand mechanistics Two new papers on G protein-coupled receptor (GPCR) structure shed new light on how different ligands interact with their cognate GPCRs. -
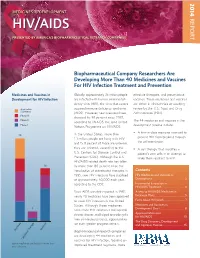
14-258 Phrma HIV/AIDS2014 0819.Indd
2014 MEDICINES IN DEVELOPMENT REPORT HIV/AIDS PRESENTED BY AMERICA’S BIOPHARMACEUTICAL RESEARCH COMPANIES Biopharmaceutical Company Researchers Are Developing More Than 40 Medicines and Vaccines For HIV Infection Treatment and Prevention Medicines and Vaccines in Globally, approximately 35 million people effective therapies, and preventative Development for HIV Infection are infected with human immunodefi - vaccines. These medicines and vaccines ciency virus (HIV), the virus that causes are either in clinical trials or awaiting Application acquired immune defi ciency syndrome review by the U.S. Food and Drug Submitted (AIDS). However, new infections have Administration (FDA). Phase III dropped by 38 percent since 2001, Phase II The 44 medicines and vaccines in the according to UNAIDS, the Joint United Phase I development pipeline include: Nations Programme on HIV/AIDS. • A fi rst-in-class medicine intended to In the United States, more than 25 prevent HIV from breaking through 1.1 million people are living with HIV the cell membrane. and 15.8 percent of those are unaware they are infected, according to the • A cell therapy that modifi es a U.S. Centers for Disease Control and patient’s own cells in an attempt to Prevention (CDC). Although the U.S. make them resistant to HIV. HIV/AIDS-related death rate has fallen 16 by more than 80 percent since the introduction of antiretroviral therapies in Contents 1995, new HIV infections have stabilized HIV Medicines and Vaccines in at approximately 50,000 each year, Development ......................................2 according to the CDC. Incremental Innovation in HIV/AIDS Treatment .......................... 4 Since AIDS was fi rst reported in 1981, Access to HIV/AIDS Medicines in nearly 40 medicines have been approved Exchange Plans ...................................5 to treat HIV infection in the United Facts About HIV/AIDS ........................7 States. -

Management of Hiv Infection
MANAGEMENT OF HIV INFECTION Federal Bureau of Prisons Clinical Guidance September 2018 Clinical guidance is made available to the public for informational purposes only. The Federal Bureau of Prisons (BOP) does not warrant this guidance for any other purpose, and assumes no responsibility for any injury or damage resulting from the reliance thereof. Proper medical practice necessitates that all cases are evaluated on an individual basis and that treatment decisions are patient-specific. Referenced Program Statement versions within this document are for informational purposes only. Please refer to the most current version of the referenced Program Statement. Consult the BOP Health Management Resources Web page to determine the date of the most recent update to this document: http://www.bop.gov/resources/health_care_mngmt.jsp Federal Bureau of Prisons Management of HIV Infection Clinical Guidance September 2018 WHAT’S NEW IN THIS DOCUMENT? This document updates the December 2017 version of the BOP Clinical Guidance on the Management of HIV Infection. Treatment information throughout this document was updated to be in line with the Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents issued by the Department of Health and Human Services (DHHS) in 2018. NOTEWORTHY CHANGES INCLUDE REVISIONS IN THE FOLLOWING AREAS: • INDICATIONS FOR HIV TESTING: An opt out strategy of voluntary testing for HIV infection is recommended for all inmates, regardless of sentencing status, including new intakes and those already established in the population who have not declined testing. • MENINGOCOCCAL VACCINE: Adults with HIV infection who have not been previously vaccinated should receive a 2-dose primary series of MenACWY (MCV4), available as Menactra® or Menveo®, at least 2 months apart, and then revaccinated every 5 years. -

Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology Xuanmao Jiao1, Omar Nawab1,2,Tejal Patel2, Andrew V
Published OnlineFirst July 10, 2019; DOI: 10.1158/0008-5472.CAN-19-1167 Cancer Review Research Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology Xuanmao Jiao1, Omar Nawab1,2,Tejal Patel2, Andrew V. Kossenkov3, Niels Halama4, Dirk Jaeger4,5, and Richard G. Pestell1,3 Abstract Experiments of nature have revealed the peculiar impor- reexpression augments resistance to DNA-damaging agents tance of the G-protein–coupled receptor, C-C chemokine and is sufficient to induce cancer metastasis and "stemness". receptor type 5 (CCR5), in human disease since ancient times. Recent studies suggest important cross-talk between CCR5 The resurgence of interest in heterotypic signals in the onset signaling and immune checkpoint function. Because CCR5 and progression of tumorigenesis has led to the current focus onTregsservesasthecoreceptorforhumanimmunodefi- on CCR5 as an exciting new therapeutic target for metastatic ciency virus (HIV) entry, CCR5-targeted therapeutics used in cancer with clinical trials now targeting breast and colon HIV, [small molecules (maraviroc and vicriviroc) and a cancer. The eutopic expression of CCR5 activates calcium humanized mAb (leronlimab)], are now being repositioned signaling and thereby augments regulatory T cell (Treg) dif- in clinical trials as cancer therapeutics. As CCR5 is expressed ferentiation and migration to sites of inflammation. The mis- on a broad array of tumors, the opportunity for therapeutic expression of CCR5 in epithelial cells, induced upon onco- repositioning and the rationale for combination therapy genic transformation, hijacks this migratory phenotype. CCR5 approaches are reviewed herein. C-C Chemokine Receptor Type 5 and Signal is preserved in immune cells (3) and cancer cells (4, 5). -

Targeting Cell Entry of Enveloped Viruses As an Antiviral Strategy
Molecules 2011, 16, 221-250; doi:10.3390/molecules16010221 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Targeting Cell Entry of Enveloped Viruses as an Antiviral Strategy Elodie Teissier, François Penin and Eve-Isabelle Pécheur * Institut de Biologie et Chimie des Protéines, UMR 5086, Université de Lyon, IFR 128 BioSciences Gerland-Lyon Sud, 69367 Lyon, France; E-Mails: [email protected] (E.T.); [email protected] (F.P.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Fax: +33 472 72 26 04. Received: 6 October 2010; in revised form: 16 December 2010 / Accepted: 24 December 2010 / Published: 30 December 2010 Abstract: The entry of enveloped viruses into their host cells involves several successive steps, each one being amenable to therapeutic intervention. Entry inhibitors act by targeting viral and/or cellular components, through either the inhibition of protein-protein interactions within the viral envelope proteins or between viral proteins and host cell receptors, or through the inhibition of protein-lipid interactions. Interestingly, inhibitors that concentrate into/onto the membrane in order to target a protein involved in the entry process, such as arbidol or peptide inhibitors of the human immunodeficiency virus (HIV), could allow the use of doses compatible with therapeutic requirements. The efficacy of these drugs validates entry as a point of intervention in viral life cycles. Strategies based upon small molecule antiviral agents, peptides, proteins or nucleic acids, would most likely prove efficient in multidrug combinations, in order to inhibit several steps of virus life cycle and prevent disease progression. -

Antiretroviral Drugs Impact Autophagy with Toxic Outcomes
cells Review Antiretroviral Drugs Impact Autophagy with Toxic Outcomes Laura Cheney 1,*, John M. Barbaro 2 and Joan W. Berman 2,3 1 Division of Infectious Diseases, Department of Medicine, Montefiore Medical Center and Albert Einstein College of Medicine, 1300 Morris Park Ave, Bronx, NY 10461, USA 2 Department of Pathology, Montefiore Medical Center and Albert Einstein College of Medicine, 1300 Morris Park Ave, Bronx, NY 10461, USA; [email protected] (J.M.B.); [email protected] (J.W.B.) 3 Department of Microbiology and Immunology, Montefiore Medical Center and Albert Einstein College of Medicine, 1300 Morris Park Ave, Bronx, NY 10461, USA * Correspondence: [email protected]; Tel.: +1-718-904-2587 Abstract: Antiretroviral drugs have dramatically improved the morbidity and mortality of peo- ple living with HIV (PLWH). While current antiretroviral therapy (ART) regimens are generally well-tolerated, risks for side effects and toxicity remain as PLWH must take life-long medications. Antiretroviral drugs impact autophagy, an intracellular proteolytic process that eliminates debris and foreign material, provides nutrients for metabolism, and performs quality control to maintain cell homeostasis. Toxicity and adverse events associated with antiretrovirals may be due, in part, to their impacts on autophagy. A more complete understanding of the effects on autophagy is essential for developing antiretroviral drugs with decreased off target effects, meaning those unrelated to viral suppression, to minimize toxicity for PLWH. This review summarizes the findings and highlights the gaps in our knowledge of the impacts of antiretroviral drugs on autophagy. Keywords: HIV; antiretroviral drugs; side effects; toxicity; autophagy; mitophagy; mitochondria; Citation: Cheney, L.; Barbaro, J.M.; ER stress Berman, J.W. -

SARS-Cov-2: an Update on Potential Antivirals in Light of SARS-Cov Antiviral Drug Discoveries
Review SARS-CoV-2: An Update on Potential Antivirals in Light of SARS-CoV Antiviral Drug Discoveries Hatem A. Elshabrawy Department of Molecular and Cellular Biology, College of Osteopathic Medicine, Sam Houston State University, Conroe, TX 77304, USA; [email protected] Received: 18 May 2020; Accepted: 17 June 2020; Published: 23 June 2020 Abstract: Coronaviruses (CoVs) are a group of RNA viruses that are associated with different diseases in animals, birds, and humans. Human CoVs (HCoVs) have long been known to be the causative agents of mild respiratory illnesses. However, two HCoVs associated with severe respiratory diseases are Severe Acute Respiratory Syndrome-CoV (SARS-CoV) and Middle East Respiratory Syndrome-CoV (MERS-CoV). Both viruses resulted in hundreds of deaths after spreading to several countries. Most recently, SARS-CoV-2 has emerged as the third HCoV causing severe respiratory distress syndrome and viral pneumonia (known as COVID-19) in patients from Wuhan, China, in December 2019. Soon after its discovery, SARS-CoV-2 spread to all countries, resulting in millions of cases and thousands of deaths. Since the emergence of SARS-CoV, many research groups have dedicated their resources to discovering effective antivirals that can treat such life-threatening infections. The rapid spread and high fatality rate of SARS-CoV-2 necessitate the quick discovery of effective antivirals to control this outbreak. Since SARS-CoV-2 shares 79% sequence identity with SARS-CoV, several anti-SARS-CoV drugs have shown promise in limiting SARS-CoV-2 replication in vitro and in vivo. In this review, we discuss antivirals described for SARS-CoV and provide an update on therapeutic strategies and antivirals against SARS-CoV-2. -

Antiviral Research and Development Against Dengue Virus
Antiviral Research and Development Against Dengue Virus Bruno Canard, PhD. [email protected] 1 Table of Contents Part 1. Antivirals 3 A short historical view on antiviral research and therapies 3 Lessons learned from recent viral diseases and pandemies 3 The methods used to discover antivirals 4 Infected cell assays 5 Knowledge-based methods 5 The source of anti-infectious molecules 6 Why has natural product screening been neglected in antiviral research ? 8 Challenges associated with natural products in antiviral research 8 What is a validated antiviral target ? 9 Animal models 9 Patient cohorts and clinical trials 10 Frequent arguments about antiviral therapy feasibility 10 The introduction of dengue as a druggable disease 11 Diagnostics, and what does it tells us for antiviral therapy ? 11 Current treatment 11 Part 2. Dengue 13 Preamble 13 The Dengue Virus 13 The DENV targets for antiviral research 13 Overview of genome organisation 14 Overview of the DV particle and DV proteins as targets for drugs 14 The structural proteins 14 The Non-Structural proteins 15 RNA structures 17 The dengue validated targets 17 The cellular targets for antiviral research against dengue 18 siRNAs as tools and/or therapeutic agents 19 Response modifiers 20 Monoclonal antibodies 21 Mechanical devices 21 Part 3. Academic and academy-associated research centers 22 Part 4. The current industrial network of AV discovery 31 Part 5. Mapping the dengue drug design effort and needs 38 Annex 1. References 42 Annex 2. Patents 43 2 Part 1. Antivirals A short historical view on antiviral research and therapies The first significant successes of anti-infectious disease treatments originated from the discovery and use of antibiotics. -

How Entry Inhibitors Work
How Entry Inhibitors Work OVERVIEW HIV attacks cells within the body’s immune system. To spread, the virus needs to enter these cells and make copies of itself. The copies are then released from these cells and infect other cells. Treatment with entry inhibitors is one way to help stop the virus from replicating and control HIV infection. Entry inhibitors work by preventing HIV from entering healthyCD4 cells (T-cells) in the body. They work differently than many other ARVs—nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), and protease inhibitors (PIs)—which are active against HIVafter it has infected a CD4 cell. Entry inhibitors work by attaching themselves to proteins on the surface of CD4 cells or proteins on the surface of HIV. In order for HIV to bind to CD4 cells, the proteins on HIV's outer coat must bind to the proteins on the surface of CD4 cells. Entry inhibitors prevent this from happening. There are different types of entry inhibitors—fusion inhibitors, receptor blockers (CCR5 antagonists), and post- attachment inhibitors. Some entry inhibitors target the gp120 or gp41 proteins on HIV's surface. Some entry inhibitors target the CD4 protein or the CCR5 or CXCR4 receptors on a CD4 cell's surface. If entry inhibitors are successful in blocking these proteins, HIV is unable to bind to the surface of CD4 cells and gain entry into the cells. People with HIV who have become resistant to NRTIs, NNRTIs, and PIs will likely benefit from entry inhibitors because they are a different class of drugs. -

Full PDF of 2003 Pipeline Report
Antiretrovirals Pipeline Report prepared for Treatment Action Group by Rob Camp Table of contents 1. New agents chart..............................................................................................................2 Extracellular agents ............................................................................................................3 NRTIs ................................................................................................................................. 11 NNRTIs .............................................................................................................................. 15 Transcriptase and Integrase inhibitors.......................................................................... 19 Protease inhibitors ........................................................................................................... 21 Budding/maturation inhibitors......................................................................................... 33 Antivirals Pipeline Report The pipeline is bursting, with at least the following compounds twinkling in the eyes of scientists around the globe. It is by no means a full list, but those substances in red were presented at the recent CROI in Boston. There seems to be no lack of investigation — bench scientists are having a field day! Compound Class of Compound Phase of Pharmaceutical Co. Development TAK 220 CCR5 antagonist Preclinical Takeda PRO 140 CCR5 antagonist Preclinical Progenics AK 602 CCR5 antagonist Preclinical Ono/Moravek SCH D CCR5 antagonist -

Adverse Effects of Antiretroviral Medications
© National HIV Curriculum PDF created October 6, 2021, 3:01 am Adverse Effects of Antiretroviral Medications This is a PDF version of the following document: Module 3: Antiretroviral Therapy Lesson 2: Adverse Effects of Antiretroviral Medications You can always find the most up to date version of this document at https://www.hiv.uw.edu/go/antiretroviral-therapy/adverse-effects/core-concept/all. Introduction Background Antiretroviral therapy has transformed HIV into a manageable chronic disease, but antiretroviral medications have the potential to cause short-term and long-term adverse effects. Medication-related adverse effects may manifest in overt symptoms or initially only as laboratory abnormalities.[1] The spectrum of potential antiretroviral drug toxicity is broad, including renal toxicity, mitochondrial and metabolic effects, gastrointestinal symptoms, weight gain, cardiovascular effects, hypersensitivity, skin reactions, insomnia, and neuropsychiatric manifestations. In general, newer antiretroviral medications have improved safety profiles compared with older antiretroviral medications, and this is reflected in the recommendations issued in the Adult and Adolescent ARV Guidelines.[2] Clinicians who provide care to persons with HIV should have an understanding of the basic toxicity profile of antiretroviral medications, keeping in mind that the potential adverse effects of most antiretroviral medications are less toxic than the effects of untreated HIV. This Topic Review will explore antiretroviral-associated adverse effects by drug class and by specific drug. Issues related to drug interactions with antiretroviral medications are addressed in the Topic Review Drug Interactions with Antiretroviral Therapy. Safety Laboratory Monitoring in Persons Taking Antiretroviral Therapy All persons with HIV who initiate antiretroviral therapy should have laboratory studies performed at the initial visit, before initiating or changing a regimen, and as regular monitoring for long-term safety once a regimen is initiated.