Triumeq, INN-Dolutegravir, Abacavir, Lamivudine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
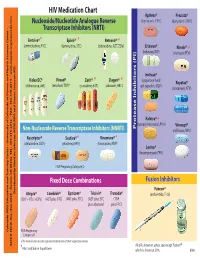
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (Tipranavir, TPV) (Darunavir, DRV) Transcriptase Inhibitors (NRTI)
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (tipranavir, TPV) (darunavir, DRV) Transcriptase Inhibitors (NRTI) Emtriva®* Epivir® * Retrovir® * (emtricitabine, FTC) (lamivudine, 3TC) (zidovudine, AZT, ZDV) Crixivan® Norvir® * (indinavir, IDV) (ritonavir, RTV) Invirase® Videx EC® Viread® Zerit® Ziagen® * * (saquinavir hard Reyataz® (didanosine, ddl) (tenofovir,TDF)* (stavudine, d4T) (abacavir, ABC) gel capsules, SQV) (atazanavir, ATV) Kaletra® * (lopinavir/ritonavir, LPV/r) Viracept® Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) Inhibitors (PI) Protease (nelfinavir, NFV) Rescriptor® Sustiva®* Viramune® * (delavirdine, DLV) (efavirenz, EFV) (nevirapine, NVP) Lexiva® (fosamprenavir, FPV) FDA Pregnancy Category D Fixed Dose Combinations Fusion Inhibitors Fuzeon® Developed by MeriLou Johnson, MeriLou by Developed Johnson, MSW, and Steven MPA MD Atripla® Combivir® Epzicom® Trizivir® Truvada® (enfuvirtide,T-20) (TDF+FTC+EFV) (AZT plus 3TC) (ABC plus 3TC) (AZT plus 3TC (TDF plus abacavir) plus FTC) Colorado AIDS Education and Training Center • University of Colorado at Denver and Health Sciences Center • Center and Health Sciences Denver at of Colorado • University Center Training and AIDS Education Colorado FDA Pregnancy Category D ®The brands listed are the registered trademarks of their respective owners. 4200 E. Ave., Ninth TEL: A089 • Denver, Box 80262 CO 303-315-2512 • FAX: • 303-315-2514 • www.mpaetc.org/colorado.htm All pills shown in actual size except Fuzeon® *Also available in liquid form. which is shown at 50%. 8/06 Medication Schedule Name______________________________ Date___________ Number of Time of day you ar to take TOTAL Name of Medication pills to take this medicine Food Interactions Side Effects number of each time pills each day With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ Discontinued Medications or Formulations Helpful Hints: • Refill prescriptions before you run out. -

Targeting Fibrosis in the Duchenne Muscular Dystrophy Mice Model: an Uphill Battle
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427485; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title: Targeting fibrosis in the Duchenne Muscular Dystrophy mice model: an uphill battle 2 Marine Theret1#, Marcela Low1#, Lucas Rempel1, Fang Fang Li1, Lin Wei Tung1, Osvaldo 3 Contreras3,4, Chih-Kai Chang1, Andrew Wu1, Hesham Soliman1,2, Fabio M.V. Rossi1 4 1School of Biomedical Engineering and the Biomedical Research Centre, Department of Medical 5 Genetics, 2222 Health Sciences Mall, Vancouver, BC, V6T 1Z3, Canada 6 2Department of Pharmacology and Toxicology, Faculty of Pharmaceutical Sciences, Minia 7 University, Minia, Egypt 8 3Developmental and Stem Cell Biology Division, Victor Chang Cardiac Research Institute, 9 Darlinghurst, NSW, 2010, Australia 10 4Departamento de Biología Celular y Molecular and Center for Aging and Regeneration (CARE- 11 ChileUC), Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, 8331150 12 Santiago, Chile 13 # Denotes Co-first authorship 14 15 Keywords: drug screening, fibro/adipogenic progenitors, fibrosis, repair, skeletal muscle. 16 Correspondence to: 17 Marine Theret 18 School of Biomedical Engineering and the Biomedical Research Centre 19 University of British Columbia 20 2222 Health Sciences Mall, Vancouver, British Columbia 21 Tel: +1(604) 822 0441 fax: +1(604) 822 7815 22 Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427485; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Viiv Healthcare Drug Class1,4: Antiretroviral Agent, Integrase
Brand Name: Tivicay Generic Name: dolutegravir Manufacturer1: ViiV Healthcare Drug Class1,4: Antiretroviral Agent, Integrase Inhibitor Labeled Uses4,5: Labeled1,4: In combination with other antiretroviral agents for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and children aged 12 years and older and weighing at least 40 kg. Mechanism of Action1,2: Dolutegravir inhibits the catalytic activity of HIV integrase, which is an HIV encoded enzyme required for viral replication. Integrase is one of the three HIV-1 enzymes required for viral replication. Integration of HIV into cellular DNA is a multi-step process. First, the assembly of integrase in a stable complex with the viral DNA occurs. Second, the terminal dinucleotides from each end of the viral DNA are removed by endonucleolytic processing. Lastly, the viral DNA 3' ends are covalently linked to the cellular (target) DNA by strand transfer. The last two processes, which are catalytic, require integrase to be appropriately assembled on a specific viral DNA substrate. Inhibition of integrase by dolutegravir prevents the covalent insertion, or integration, of unintegrated linear HIV DNA into the host cell genome preventing the formation of the HIV provirus. The provirus is required to direct the production of progeny virus, so inhibiting integration prevents propagation of the viral infection. Pharmacokinetics1: Absorption: Tmax 2-3 hours Vd 17.4L T1/2 14 hours Clearance 1.0 L/h Protein Binding >98.9% Bioavailability Not established Metabolism1,2: Dolutegravir is primarily metabolized via UGT1A1 with some contribution from CYP3A. Metabolism occurs via UDP-glucuronosyltransferase (UGT)1A1 (major) and by the hepatic isoenzyme CYP3A (minor). -

HIV Integrase Inhibitor Pharmacogenetics and Clinical Outcomes: an Exploratory Association Study Derek E
East Tennessee State University Digital Commons @ East Tennessee State University Electronic Theses and Dissertations Student Works 8-2018 HIV Integrase Inhibitor Pharmacogenetics and Clinical Outcomes: An Exploratory Association Study Derek E. Murrell East Tennessee State University Follow this and additional works at: https://dc.etsu.edu/etd Part of the Other Pharmacy and Pharmaceutical Sciences Commons, Pharmacology Commons, and the Virus Diseases Commons Recommended Citation Murrell, Derek E., "HIV Integrase Inhibitor Pharmacogenetics and Clinical Outcomes: An Exploratory Association Study" (2018). Electronic Theses and Dissertations. Paper 3465. https://dc.etsu.edu/etd/3465 This Dissertation - Open Access is brought to you for free and open access by the Student Works at Digital Commons @ East Tennessee State University. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons @ East Tennessee State University. For more information, please contact [email protected]. HIV Integrase Inhibitor Pharmacogenetics and Clinical Outcomes: An Exploratory Association Study _____________________ A dissertation presented to the faculty of the Department of Biomedical Sciences East Tennessee State University In partial fulfillment of the requirements for the degree Doctor of Philosophy in Biomedical Sciences, Pharmaceutical Sciences Concentration _____________________ by Derek Edward Murrell August 2018 _____________________ Sam Harirforoosh, PharmD, PhD, Chair Jonathan Moorman, MD, PhD David Roane, PhD Robert Schoborg, PhD Zhi Qiang Yao, MD, PhD Keywords: Integrase Strand Transfer Inhibitor, Dolutegravir, Elvitegravir, Raltegravir, Pharmacogenetics, HIV, Renal, Hepatic, Adverse events ABSTRACT HIV Integrase Inhibitor Pharmacogenetics and Clinical Outcomes: An Exploratory Association Study by Derek Edward Murrell As HIV is now primarily a chronic condition, treatment is given life-long with changes as necessitated by alterations in tolerability and efficacy. -

Product Monograph for CELSENTRI
PRODUCT MONOGRAPH PrCELSENTRI maraviroc Tablets 150 and 300 mg CCR5 antagonist ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Revision: July 05, 2019 Submission Control No: 226222 © 2019 ViiV Healthcare group of companies or its licensor. Trademarks are owned by or licensed to the ViiV Healthcare group of companies. Page 1 of 60 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION.........................................................3 SUMMARY PRODUCT INFORMATION ........................................................................3 INDICATIONS AND CLINICAL USE..............................................................................3 CONTRAINDICATIONS ...................................................................................................3 WARNINGS AND PRECAUTIONS..................................................................................4 ADVERSE REACTIONS....................................................................................................9 DRUG INTERACTIONS ..................................................................................................19 DOSAGE AND ADMINISTRATION..............................................................................28 OVERDOSAGE ................................................................................................................31 ACTION AND CLINICAL PHARMACOLOGY ............................................................31 STORAGE AND STABILITY..........................................................................................36 -

Recommendations on First and Second Line Antiretroviral Regimens
POLICY BRIEF UPDATE OF RECOMMENDATIONS ON FIRST- AND SECOND-LINE ANTIRETROVIRAL REGIMENS JULY 2019 HIV TREATMENT WHO/CDS/HIV/19.15 © World Health Organization 2019 Some rights reserved. This work is available under the Creative Commons Attribution- NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/ licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. Update of recommendations on first- and second-line antiretroviral regimens. Geneva, Switzerland: World Health Organization; 2019 (WHO/CDS/HIV/19.15). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. CIP data are available at http://apps.who.int/iris. Sales, rights and licensing. -

Non-Anntoated Product Monograph
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION PrTRIUMEQ dolutegravir, abacavir, and lamivudine tablets 50 mg dolutegravir (as dolutegravir sodium), 600 mg abacavir (as abacavir sulfate) and 300 mg lamivudine Antiretroviral Agent ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Revision: January 31, 2020 Submission Control No: 233245 © 2020 ViiV Healthcare group of companies or its licensor Trademarks are owned by or licensed to the ViiV Healthcare group of companies Page 1 of 61 TABLE OF CONTENTS PAGE PART I: HEALTH PROFESSIONAL INFORMATION ........................................................ 3 SUMMARY PRODUCT INFORMATION ................................................................... 3 INDICATIONS AND CLINICAL USE ........................................................................ 3 CONTRAINDICATIONS ........................................................................................... 4 WARNINGS AND PRECAUTIONS............................................................................ 4 ADVERSE REACTIONS.......................................................................................... 11 DRUG INTERACTIONS .......................................................................................... 19 DOSAGE AND ADMINISTRATION ........................................................................ 24 OVERDOSAGE....................................................................................................... 26 ACTION AND CLINICAL PHARMACOLOGY........................................................ -

Selected Properties of Maraviroc
Selected Properties of Maraviroc Other names UK-427,857, MVC, Celsentri , Selzentry (US) Manufacturer ViiV Healthcare ULC Pharmacology/Mechanism of Maraviroc is a selective, slowly reversible, small molecule Action antagonist of the interaction between human CCR5 and HIV-1 gp120. Blocking this interaction prevents CCR5-tropic HIV-1 entry into cells . CCR5 antagonists target a discrete step in the viral entry pathway. The mechanism of HIV entry into the host CD4 T cells involves a sequence of molecular interactions between the virion envelope glycoprotein (Env) and host cell surface receptors. Normally, the gp120 Env subunit binds to CD4, and subsequent binding of HIV to the host cell’s coreceptors (CCR5 or CXCR4) causes a conformational change leading to membrane fusion into the host cell. Allosteric binding of a CCR5 antagonist results in a receptor conformation that the virus cannot bind to, thus interfering with the fusion process. NB: Use of maraviroc is not recommended in patients with dual/mixed or CXCR4-tropic HIV-1 as efficacy was not demonstrated in a phase 2 study of this patient group. The mean EC value (50% effective concentration) for Activity 50 maraviroc against HIV-1 group M isolates (clades A to J) and group O isolates ranged from 0.1 to 1.25 nM (0.05 to 0.64 ng/mL) in cell culture. Mean potency against a range of CCR5- tropic clinical primary isolates: IC 90 2.03 nM (1.04 ng/mL). In 973 treatment-experienced HIV-1-infected subjects in studies A4001027 and A4001028, the C , baseline viral load, baseline min CD4, cell count and overall sensitivity score (OSS) were found to be important predictors of virologic success (defined as viral load < 400 copies/mL at 24 weeks). -

(12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson Et Al
US 20080241135A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson et al. (43) Pub. Date: Oct. 2, 2008 (54) METHODS FOR REDUCINGVIRAL LOAD IN 60/711,528, filed on Aug. 26, 2005, provisional appli HIV-1-INFECTED PATIENTS cation No. 60/715,619, filed on Sep. 9, 2005. (75) Inventors: William C. Olson, Ossining, NY Publication Classification (US); Paul J. Maddon, Scarsdale, NY (US); Daniel C. Pevear, (51) Int. Cl. Medford, MA (US); Robert J. A 6LX 39/395 (2006.01) Israel, Suffern, NY (US); Jose D. A6IP 43/00 (2006.01) Murga, Rosedale, NY (US) (52) U.S. Cl. ..................................................... 424/133.1 Correspondence Address: (57) ABSTRACT COOPER & DUNHAM, LLP 1185 AVENUE OF THE AMERICAS This method provides a method for reducing HIV-1 viral load NEW YORK, NY 10036 in an HIV-1-infected human subject which comprises admin istering to the subject at a predefined interval effective HIV-1 (73) Assignee: Progenics Pharmaceuticals, Inc. viral load-reducing doses of (a) a humanized antibody desig nated PRO 140, or of (b) an anti-CCR5 receptor monoclonal (21) Appl. No.: 11/894,762 antibody. This invention also provides a method for inhibiting in a human Subject the onset or progression of an HIV-1- (22) Filed: Aug. 20, 2007 associated disorder, the inhibition of which is effected by inhibiting fusion of HIV-1 to CCR5"CD4" target cells in the Related U.S. Application Data Subject. This invention also provides a method for treating a subject infected with HIV-1 comprising administering to the (63) Continuation of application No. -

Of 43 Reference ID: 4711890 FULL PRESCRIBING INFORMATION: CONTENTS*
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOLUTEGRAVIR, EMTRICITABINE and TENOFOVIR -----------------------WARNINGS AND PRECAUTIONS------------------------ ALAFENAMIDE TABLETS safely and effectively. See full prescribing • Hypersensitivity reactions characterized by rash, constitutional findings, information for DOLUTEGRAVIR, EMTRICITABINE and and sometimes organ dysfunction, including liver injury have been TENOFOVIR ALAFENAMIDE TABLETS. reported. Discontinue dolutegravir, emtricitabine and tenofovir alafenamide tablets and other suspect agents immediately if signs or DOLUTEGRAVIR, EMTRICITABINE and TENOFOVIR symptoms of hypersensitivity reactions develop. (5.2) ALAFENAMIDE tablets, for oral use • Hepatotoxicity has been reported in patients receiving a dolutegravir- containing regimen. Monitoring for hepatotoxicity is recommended. WARNING: POST-TREATMENT ACUTE EXACERBATION OF (5.3) HEPATITIS B • Embryo-fetal toxicity may occur when used at the time of conception and in early pregnancy. An alternative treatment to dolutegravir, See full prescribing information for complete boxed warning. emtricitabine and tenofovir alafenamide tablets should be considered at the time of conception through the first trimester of pregnancy due to the Severe acute exacerbations of hepatitis B virus (HBV) have been reported risk of neural tube defects. Counsel adolescents and adults of in HBV-infected patients who have discontinued products containing childbearing potential to use effective contraception. -

Etravirine (Intelence)
etravirine (Intelence) WHAT IS ETRAVIRINE? Etravirine, also known as ETR (brand name Intelence), is a drug used as part ofantiretroviral therapy (ART). The FDA approved etravirine in 2008 as an antiretroviral drug (ARV) for people with HIV infection. Etravirine is manufactured by Janssen Therapeutics. Etravirine is a type of drug called a non-nucleoside reverse transcriptase inhibitor (NNRTI). NNRTIs bind to and block reverse transcriptase (an HIV enzyme). HIV uses reverse transcriptase to convert its RNA into DNA (reverse transcription). Blocking reverse transcriptase and reverse transcription prevents HIV from replicating. When used in combination with other ARVs to treat HIV infection, etravirine may help: Reduce the amount of HIV in your blood. This is called viral load. Increase the number of CD4 cells in your blood that help fight off other infections. Reducing the amount of HIV and increasing the CD4 cells in your blood may help improve your immune system. This may reduce your risk of death or gettingopportunistic infections (OIs) that can happen when your immune system is weak. Read more about viral suppression. Etravirine does not cure HIV infection or AIDS. You must keep taking HIV medicines to control HIV infection and decrease HIV-related illnesses. WHO SHOULD TAKE ETRAVIRINE? Etravirine is a prescription HIV medicine used in combination with other ARVs to treat HIV infection in adults and children 2 years of age and older who have taken HIV medicines in the past. The safety and effectiveness of etravirine has not been established in children under 2 years of age. Etravirine has not been carefully studied in the elderly (65 years of age and older). -

Guidelines for the Use of Antiretroviral Agents in Adults and Adolescent Living With
Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV Developed by the DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents – A Working Group of the Office of AIDS Research Advisory Council (OARAC) How to Cite the Adult and Adolescent Guidelines: Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services. Available at https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/ AdultandAdolescentGL.pdf. Accessed [insert date] [insert page number, table number, etc. if applicable] It is emphasized that concepts relevant to HIV management evolve rapidly. The Panel has a mechanism to update recommendations on a regular basis, and the most recent information is available on the HIVinfo Web site (http://hivinfo.nih.gov). What’s New in the Guidelines? August 16, 2021 Hepatitis C Virus/HIV Coinfection • Table 18 of this section has been updated to include recommendations regarding concomitant use of fostemsavir or long acting cabotegravir plus rilpivirine with different hepatitis C treatment regimens. June 3, 2021 What to Start • Since the release of the last guidelines, updated data from the Botswana Tsepamo study have shown that the prevalence of neural tube defects (NTD) associated with dolutegravir (DTG) use during conception is much lower than previously reported. Based on these new data, the Panel now recommends that a DTG-based regimen can be prescribed for most people with HIV who are of childbearing potential. Before initiating a DTG-based regimen, clinicians should discuss the risks and benefits of using DTG with persons of childbearing potential, to allow them to make an informed decision.