Odefsey Gilead Sciences Pty Ltd PM-2015-02479-1-2 Final 12 October 2017
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Emtricitabine, Rilpivirine, and Tenofovir Disoproxil Fumarate
PATIENT & CAREGIVER EDUCATION Emtricitabine, Rilpivirine, and Tenofovir Disoproxil Fumarate This information from Lexicomp® explains what you need to know about this medication, including what it’s used for, how to take it, its side effects, and when to call your healthcare provider. Brand Names: US Complera Brand Names: Canada Complera Warning Hepatitis B has gotten worse when this drug was stopped in some people with hepatitis B. Close follow-up for a few months is needed when therapy is stopped in people who have hepatitis B. Do not stop taking this drug without calling your doctor. Hepatitis B testing needs to be done as you were told by the doctor. Talk with the doctor. What is this drug used for? It is used to treat HIV infection. Emtricitabine, Rilpivirine, and Tenofovir Disoproxil Fumarate 1/7 What do I need to tell my doctor BEFORE I take this drug? If you are allergic to this drug; any part of this drug; or any other drugs, foods, or substances. Tell your doctor about the allergy and what signs you had. If you have kidney disease. If you take any drugs (prescription or OTC, natural products, vitamins) that must not be taken with this drug, like some other drugs used for HIV or certain drugs used for seizures, infection, or stomach or bowel problems. There are many drugs that must not be taken with this drug. If you are breast-feeding. Do not breast-feed while you take this drug. This is not a list of all drugs or health problems that interact with this drug. -
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (Tipranavir, TPV) (Darunavir, DRV) Transcriptase Inhibitors (NRTI)
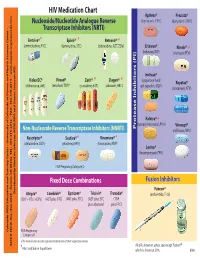
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (tipranavir, TPV) (darunavir, DRV) Transcriptase Inhibitors (NRTI) Emtriva®* Epivir® * Retrovir® * (emtricitabine, FTC) (lamivudine, 3TC) (zidovudine, AZT, ZDV) Crixivan® Norvir® * (indinavir, IDV) (ritonavir, RTV) Invirase® Videx EC® Viread® Zerit® Ziagen® * * (saquinavir hard Reyataz® (didanosine, ddl) (tenofovir,TDF)* (stavudine, d4T) (abacavir, ABC) gel capsules, SQV) (atazanavir, ATV) Kaletra® * (lopinavir/ritonavir, LPV/r) Viracept® Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) Inhibitors (PI) Protease (nelfinavir, NFV) Rescriptor® Sustiva®* Viramune® * (delavirdine, DLV) (efavirenz, EFV) (nevirapine, NVP) Lexiva® (fosamprenavir, FPV) FDA Pregnancy Category D Fixed Dose Combinations Fusion Inhibitors Fuzeon® Developed by MeriLou Johnson, MeriLou by Developed Johnson, MSW, and Steven MPA MD Atripla® Combivir® Epzicom® Trizivir® Truvada® (enfuvirtide,T-20) (TDF+FTC+EFV) (AZT plus 3TC) (ABC plus 3TC) (AZT plus 3TC (TDF plus abacavir) plus FTC) Colorado AIDS Education and Training Center • University of Colorado at Denver and Health Sciences Center • Center and Health Sciences Denver at of Colorado • University Center Training and AIDS Education Colorado FDA Pregnancy Category D ®The brands listed are the registered trademarks of their respective owners. 4200 E. Ave., Ninth TEL: A089 • Denver, Box 80262 CO 303-315-2512 • FAX: • 303-315-2514 • www.mpaetc.org/colorado.htm All pills shown in actual size except Fuzeon® *Also available in liquid form. which is shown at 50%. 8/06 Medication Schedule Name______________________________ Date___________ Number of Time of day you ar to take TOTAL Name of Medication pills to take this medicine Food Interactions Side Effects number of each time pills each day With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ Discontinued Medications or Formulations Helpful Hints: • Refill prescriptions before you run out. -

Truvada (Emtricitabine / Tenofovir Disoproxil)
Pre-exposure Prophylaxis (2.3) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use Recommended dose in HIV-1 uninfected adults: One tablet TRUVADA safely and effectively. See full prescribing information (containing 200 mg/300 mg of emtricitabine and tenofovir for TRUVADA. disoproxil fumarate) once daily taken orally with or without food. (2.3) TRUVADA® (emtricitabine/tenofovir disoproxil fumarate) tablets, for oral use Recommended dose in renally impaired HIV-uninfected Initial U.S. Approval: 2004 individuals: Do not use TRUVADA in HIV-uninfected individuals if CrCl is below 60 mL/min. If a decrease in CrCl is observed in WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH uninfected individuals while using TRUVADA for PrEP, evaluate STEATOSIS, POST-TREATMENT ACUTE EXACERBATION OF potential causes and re-assess potential risks and benefits of HEPATITIS B, and RISK OF DRUG RESISTANCE WITH USE OF continued use. (2.4) TRUVADA FOR PrEP IN UNDIAGNOSED HIV-1 INFECTION -----------------------DOSAGE FORMS AND STRENGTHS------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 Lactic acidosis and severe hepatomegaly with steatosis, mg/150 mg of emtricitabine and tenofovir disoproxil fumarate . (3) including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, a component of TRUVADA. (5.1) --------------------------------CONTRAINDICATIONS----------------------------- TRUVADA is not approved for the treatment of chronic Do not use TRUVADA for pre-exposure prophylaxis in individuals with hepatitis B virus (HBV) infection. Severe acute unknown or positive HIV-1 status. TRUVADA should be used in exacerbations of hepatitis B have been reported in patients HIV-infected patients only in combination with other antiretroviral coinfected with HIV-1 and HBV who have discontinued agents. -

Download Article PDF/Slides
New Antiretrovirals in Development: Reprinted from The PRN Notebook,™ june 2002. Dr. James F. Braun, Editor-in-Chief. Tim Horn, Executive Editor. Published in New York City by the Physicians’ Research Network, Inc.,® John Graham Brown, Executive Director. For further information and other articles The View in 2002 available online, visit http://www.PRN.org All rights reserved. © june 2002. Roy “Trip” Gulick, md, mph Associate Professor of Medicine, Weill Medical College of Cornell University Director, Cornell Clinical Trials Unit, New York, New York Summary by Tim Horn Edited by Scott Hammer, md espite the fact that 16 antiretro- tiviral activity of emtricitabine was estab- Preliminary results from two random- virals are approved for use in the lished, with total daily doses of 200 mg or ized studies—FTC-302 and FTC-303—were United States, there is an indis- more producing the greatest median viral reported by Dr. Charles van der Horst and putable need for new anti-hiv com- load suppression: 1.72-1.92 log. Based on his colleagues at the 8th croi, held in Feb- pounds that have potent and these data, a once-daily dose of 200 mg ruary 2001 in Chicago (van der Horst, durable efficacy profiles, unique re- was selected for further long-term clinical 2001). FTC-302 was a blinded comparison sistance patterns, patient-friendly dosing study. “This is what we’re looking forward of emtricitabine and lamivudine, both in schedules, and minimal toxicities. To pro- to with emtricitabine,” commented Dr. combination with stavudine (Zerit) and vide prn with a glimpse of drugs current- Gulick. -

Product Monograph for CELSENTRI
PRODUCT MONOGRAPH PrCELSENTRI maraviroc Tablets 150 and 300 mg CCR5 antagonist ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Revision: July 05, 2019 Submission Control No: 226222 © 2019 ViiV Healthcare group of companies or its licensor. Trademarks are owned by or licensed to the ViiV Healthcare group of companies. Page 1 of 60 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION.........................................................3 SUMMARY PRODUCT INFORMATION ........................................................................3 INDICATIONS AND CLINICAL USE..............................................................................3 CONTRAINDICATIONS ...................................................................................................3 WARNINGS AND PRECAUTIONS..................................................................................4 ADVERSE REACTIONS....................................................................................................9 DRUG INTERACTIONS ..................................................................................................19 DOSAGE AND ADMINISTRATION..............................................................................28 OVERDOSAGE ................................................................................................................31 ACTION AND CLINICAL PHARMACOLOGY ............................................................31 STORAGE AND STABILITY..........................................................................................36 -

Reference ID: 2998411
• New onset or worsening renal impairment: Can include acute HIGHLIGHTS OF PRESCRIBING INFORMATION renal failure and Fanconi syndrome. Assess creatinine clearance These highlights do not include all the information needed to use (CrCl) before initiating treatment with COMPLERA. Monitor CrCl COMPLERA safely and effectively. See full prescribing and serum phosphorus in patients at risk. Avoid administering information for COMPLERA. COMPLERA with concurrent or recent use of nephrotoxic drugs. (5.3) COMPLERATM (emtricitabine/rilpivirine/tenofovir disoproxil • Caution should be given to prescribing COMPLERA with drugs fumarate) tablets that may reduce the exposure of rilpivirine. (5.4) Initial U.S. Approval: 2011 • Caution should be given to prescribing COMPLERA with drugs with a known risk of Torsade de Pointes. (5.4) WARNINGS: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS and POST TREATMENT ACUTE • Depressive disorders: Severe depressive disorders (depressed EXACERBATION OF HEPATITIS B mood, depression, dysphoria, major depression, mood altered, negative thoughts, suicide attempt, suicidal ideation) have been See full prescribing information for complete boxed warning. reported. Immediate medical evaluation is recommended for severe depressive disorders. (5.5) • Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of • Decreases in bone mineral density (BMD): Consider monitoring nucleoside analogs, including tenofovir disoproxil BMD in patients with a history of pathologic fracture -

AGENERASE® (Amprenavir) Capsules
NDA 21-007/SLR017 Page 2 PRESCRIBING INFORMATION AGENERASE® (amprenavir) Capsules PATIENT INFORMATION INCLUDED Because of the potential risk of toxicity from the large amount of the excipient, propylene glycol, contained in AGENERASE Oral Solution, that formulation is contraindicated in infants and children below the age of 4 years and certain other patient populations and should be used with caution in others. Consult the complete prescribing information for AGENERASE Oral Solution for full information. DESCRIPTION AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus (HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4- amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula: Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C. AGENERASE Capsules are available for oral administration. Each 50-mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50-mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU. -

Interactions Among Drugs for HIV & Opportunistic Infections
The New England Journal of Medicine Drug Therapy Metabolic Interactions All HIV-protease inhibitors and non-nucleoside re- verse-transcriptase inhibitors that have been approved A LASTAIR J.J. WOOD, M.D., Editor by the Food and Drug Administration (FDA), as well as those that are investigational drugs, are metabolized INTERACTIONS AMONG DRUGS by the cytochrome P-450 enzyme system, primarily FOR HIV AND OPPORTUNISTIC by the 3A4 isoform (CYP3A4), and each of these drugs may alter the metabolism of other antiretroviral INFECTIONS and concomitantly administered drugs.15 The cyto- chrome P-450 system consists of at least 11 families of STEPHEN C. PISCITELLI, PHARM.D., enzymes, classified by number, of which 3 (CYP1, AND KEITH D. GALLICANO, PH.D. CYP2, and CYP3) are important in humans.16 The RUG interactions are an important factor in families are further divided into subfamilies, denoted the treatment of patients with human im- by a capital letter (e.g., CYP3A). Individual proteins munodeficiency virus (HIV) infection. The within a subfamily, called isozymes or isoenzymes, are D 16 complexity of current drug regimens for such pa- identified by a second number (e.g., CYP3A4). tients requires that clinicians recognize and manage Drugs can be classified as cytochrome P-450 sub- drug interactions. Antiretroviral drug regimens typ- strates, inhibitors, or inducers. However, some drugs, ically consist of three or four antiretroviral drugs but such as ritonavir, nelfinavir, and efavirenz, may have may include even more. In addition, patients may re- properties of all three, depending on the specific com- ceive other drugs for supportive care, treatment of op- bination (Table 2). -

Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis
Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention Division of Tuberculosis Elimination Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis Centers for Disease Control and Prevention Office of Infectious Diseases National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention Division of Tuberculosis Elimination June 2013 This document is accessible online at http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm Suggested citation: CDC. Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis [online]. 2013. Available from URL: http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm Table of Contents Introduction 1 Methodology for Preparation of these Guidelines 2 The Role of Rifamycins in Tuberculosis Treatment 4 Managing Drug Interactions with Antivirals and Rifampin 5 Managing Drug Interactions with Antivirals and Rifabutin 9 Treatment of Latent TB Infection with Rifampin or Rifapentine 10 Treating Pregnant Women with Tuberculosis and HIV Co-infection 10 Treating Children with HIV-associated Tuberculosis 12 Co-treatment of Multidrug-resistant Tuberculosis and HIV 14 Limitations of these Guidelines 14 HIV-TB Drug Interaction Guideline Development Group 15 References 17 Table 1a. Recommendations for regimens for the concomitant treatment of tuberculosis and HIV infection in adults 21 Table 1b. Recommendations for regimens for the concomitant treatment of tuberculosis and HIV infection in children 22 Table 2a. Recommendations for co-administering antiretroviral drugs with RIFAMPIN in adults 23 Table 2b. Recommendations for co-administering antiretroviral drugs with RIFAMPIN in children 25 Table 3. Recommendations for co-administering antiretroviral drugs with RIFABUTIN in adults 26 ii Introduction Worldwide, tuberculosis is the most common serious opportunistic infection among people with HIV infection. -

(12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson Et Al
US 20080241135A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson et al. (43) Pub. Date: Oct. 2, 2008 (54) METHODS FOR REDUCINGVIRAL LOAD IN 60/711,528, filed on Aug. 26, 2005, provisional appli HIV-1-INFECTED PATIENTS cation No. 60/715,619, filed on Sep. 9, 2005. (75) Inventors: William C. Olson, Ossining, NY Publication Classification (US); Paul J. Maddon, Scarsdale, NY (US); Daniel C. Pevear, (51) Int. Cl. Medford, MA (US); Robert J. A 6LX 39/395 (2006.01) Israel, Suffern, NY (US); Jose D. A6IP 43/00 (2006.01) Murga, Rosedale, NY (US) (52) U.S. Cl. ..................................................... 424/133.1 Correspondence Address: (57) ABSTRACT COOPER & DUNHAM, LLP 1185 AVENUE OF THE AMERICAS This method provides a method for reducing HIV-1 viral load NEW YORK, NY 10036 in an HIV-1-infected human subject which comprises admin istering to the subject at a predefined interval effective HIV-1 (73) Assignee: Progenics Pharmaceuticals, Inc. viral load-reducing doses of (a) a humanized antibody desig nated PRO 140, or of (b) an anti-CCR5 receptor monoclonal (21) Appl. No.: 11/894,762 antibody. This invention also provides a method for inhibiting in a human Subject the onset or progression of an HIV-1- (22) Filed: Aug. 20, 2007 associated disorder, the inhibition of which is effected by inhibiting fusion of HIV-1 to CCR5"CD4" target cells in the Related U.S. Application Data Subject. This invention also provides a method for treating a subject infected with HIV-1 comprising administering to the (63) Continuation of application No. -

Antivirals: HIV –Cabotegravir/Rilpivirine (Cabenuva) Medical Policy No
Antivirals: HIV –Cabotegravir/rilpivirine (Cabenuva) Medical policy no. 12.10.99.AB-1 Effective Date: June 1, 2021 Related medical policies: • 12.10.99 Antivirals- HIV Combinations • 12.10.99.AA Antivirals – HIV: emtricitabine-tenofovir (Descovy) Note: New-to-market drugs included in this class based on the Apple Health Preferred Drug List (AHPDL) are non-preferred and subject to this prior authorization (PA) criteria. Non-preferred agents in this class require an inadequate response or documented intolerance due to severe adverse reaction or contraindication to at least ONE preferred regimen. If a drug within this policy receives a new indication approved by the Food and Drug Administration (FDA), medical necessity for the new indication will be determined on a case-by-case basis following FDA labeling. To see the list of the current AHPDL, please visit: https://www.hca.wa.gov/assets/billers-and-providers/apple-health-preferred-drug-list.xlsx Before authorization of Cabenuva will be approved, documentation from TheraCom specialty pharmacy showing the patient has been established on Vocabria must be included. Documentation must include patient’s name, patient’s date of birth, NDC, quantity/days supply, and date patient received the medication. TheraCom Pharmacy contact information: TheraCom Mailing Address: TheraCom VIIV Specific Team numbers (Used to locate TheraCom in e-Prescribing systems) Phone: 1-844-276-6299 TheraCom Fax: 1-833-904-1881 345 International Blvd Ste 200 Brooks, KY 40109 Background: Human immunodeficiency virus (HIV) is a single-stranded RNA retrovirus that attacks the immune system, specifically CD4+ T-helper cells, causing a progressive decrease in CD4+ T cell count and increased susceptibility of a person to infections. -

Injectable Antiretroviral Drugs: Back to the Future
viruses Review Injectable Antiretroviral Drugs: Back to the Future Marco Berruti 1 , Niccolò Riccardi 2, Diana Canetti 3,4 , Sergio Lo Caputo 5 , Lucia Taramasso 6 and Antonio Di Biagio 1,6,* 1 Infectious Diseases Unit, Department of Health Sciences (DISSAL), University of Genoa, 16132 Genoa, Italy; [email protected] 2 Department of Infectious-Tropical Diseases and Microbiology, IRCCS Sacro Cuore Don Calabria Hospital, Negrar di Valpolicella, 37024 Verona, Italy; [email protected] 3 Clinic of Infectious Diseases, IRCCS San Raffaele Scientific Institute, 20097 Milan, Italy; [email protected] 4 School of Medicine, Vita-Salute San Raffaele University, 20097 Milan, Italy 5 Clinic of Infectious Diseases, Department of Clinical and Experimental Medicine, University of Foggia, 71122 Foggia, Italy; [email protected] 6 Infectious Diseases Unit, Department of Internal Medicine, Ospedale Policlinico San Martino IRCCS, 16132 Genoa, Italy; [email protected] * Correspondence: [email protected] Abstract: Current HIV treatment regimens provide sustained virologic suppression, at least partially restore the immune system and have limited side effects; however, they do not allow viral eradication and they are burdened by daily pill intake with a life-long commitment for the people living with HIV (PHIV). Injectable agents might represent a turning point in the care of PHIV, allowing less frequent administration of antiretroviral treatment (ART), more widespread use of pre-exposure prophylaxis (PrEP) and more stable drug levels in the blood, thus increasing the odds to get closer to end the HIV pandemic. The aim of this manuscript is to give a comprehensive review of injectable antiretrovirals that have been used in the past, which are available now, will be available in the future, and their role in the treatment of HIV infection Citation: Berruti, M.; Riccardi, N.; Canetti, D.; Lo Caputo, S.; Taramasso, Keywords: ART; injectable; HIV; antiretroviral; treatment; PrEP L.; Di Biagio, A.