An Abstract of the Dissertation Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
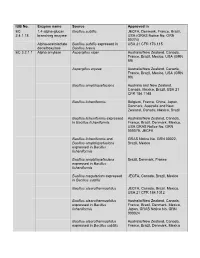
IUB No. Enzyme Name Source Approved in EC 1,4-Alpha-Glucan Bacillus Subtilis JECFA, Denmark, France, Brazil, 2.4.1.18 Branching Enzyme USA (GRAS Notice No
IUB No. Enzyme name Source Approved in EC 1,4-alpha-glucan Bacillus subtilis JECFA, Denmark, France, Brazil, 2.4.1.18 branching enzyme USA (GRAS Notice No. GRN 00274) Alpha-acetolactate Bacillus subtilis expressed in USA 21 CFR 173.115 decarboxylase Bacillus brevis EC 3.2.1.1 Alpha amylase Aspergillus niger Australia/New Zealand, Canada, France, Brazil, Mexico, USA (GRN 89) Aspergillus oryzae Australia/New Zealand, Canada, France, Brazil, Mexico, USA (GRN 90) Bacillus amyloliquefaciens Australia and New Zealand, Canada, Mexico, Brazil, USA 21 CFR 184.1148 Bacillus licheniformis Belgium, France, China, Japan, Denmark, Australia and New Zealand, Canada, Mexico, Brazil Bacillus licheniformis expressed Australia/New Zealand, Canada, in Bacillus licheniformis France, Brazil, Denmark, Mexico, USA GRAS Notice No. GRN 000079, JECFA Bacillus licheniformis and GRAS Notice No. GRN 00022, Bacillus amyloliquefaciens Brazil, Mexico expressed in Bacillus licheniformis Bacillus amyloliquefaciens Brazil, Denmark, France expressed in Bacillus licheniformis Bacillus megaterium expressed JECFA, Canada, Brazil, Mexico in Bacillus subtilis Bacillus stearothermophilus JECFA, Canada, Brazil, Mexico, USA 21 CFR 184.1012 Bacillus stearothermophilus Australia/New Zealand, Canada, expressed in Bacillus France, Brazil, Denmark, Mexico, licheniformis Japan, GRAS Notice No. GRN 000024 Bacillus stearothermophilus Australia/New Zealand, Canada, expressed in Bacillus subtilis France, Brazil, Denmark, Mexico Bacillus stearothermophilus Australia/New Zealand, Canada, (Geobacillus France, Europa, Brazil, Mexico, stearothermophilus) USA (GRN 594) Bacillus subtilis Australia and New Zealand, Canada, Mexico, Brazil, USA 21 CFR 184.1148 Rhizopus delemar Brazil Rhizopus oryzae Brazil, Mexico, USA GRAS Notice No. GRN 000090 Thermoccocales expressed in Brazil, USA (GRN126) Pseudomonas fluorecens Rhizomucor pusillus expressed Brazil, Denmark, France, Mexico, in Aspergillus niger Australia and New Zealand. -

(12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS Et Al
US 2016.0346364A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS et al. (43) Pub. Date: Dec. 1, 2016 (54) MEDICAMENT AND METHOD FOR (52) U.S. Cl. TREATING INNATE IMMUNE RESPONSE CPC ........... A61K 38/488 (2013.01); A61K 38/482 DISEASES (2013.01); C12Y 304/23019 (2013.01); C12Y 304/21026 (2013.01); C12Y 304/23018 (71) Applicant: DSM IPASSETS B.V., Heerlen (NL) (2013.01); A61K 9/0053 (2013.01); C12N 9/62 (2013.01); A23L 29/06 (2016.08); A2ID 8/042 (72) Inventors: Maaike Johanna BRUINS, Kaiseraugst (2013.01); A23L 5/25 (2016.08); A23V (CH); Luppo EDENS, Kaiseraugst 2002/00 (2013.01) (CH); Lenneke NAN, Kaiseraugst (CH) (57) ABSTRACT (21) Appl. No.: 15/101,630 This invention relates to a medicament or a dietary Supple (22) PCT Filed: Dec. 11, 2014 ment comprising the Aspergillus niger aspergilloglutamic peptidase that is capable of hydrolyzing plant food allergens, (86). PCT No.: PCT/EP2014/077355 and more particularly, alpha-amylase/trypsin inhibitors, thereby treating diseases due to an innate immune response S 371 (c)(1), in humans, and/or allowing to delay the onset of said (2) Date: Jun. 3, 2016 diseases. The present invention relates to the discovery that (30) Foreign Application Priority Data the Aspergillus niger aspergilloglutamic peptidase is capable of hydrolyzing alpha-amylase/trypsin inhibitors that are Dec. 11, 2013 (EP) .................................. 13196580.8 present in wheat and related cereals said inhibitors being strong inducers of innate immune response. Furthermore, Publication Classification the present invention relates to a method for hydrolyzing alpha-amylase/trypsin inhibitors comprising incubating a (51) Int. -

Progress in the Field of Aspartic Proteinases in Cheese Manufacturing
Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering Sirma Yegin, Peter Dekker To cite this version: Sirma Yegin, Peter Dekker. Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering. Dairy Science & Technology, EDP sciences/Springer, 2013, 93 (6), pp.565-594. 10.1007/s13594-013-0137-2. hal-01201447 HAL Id: hal-01201447 https://hal.archives-ouvertes.fr/hal-01201447 Submitted on 17 Sep 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Dairy Sci. & Technol. (2013) 93:565–594 DOI 10.1007/s13594-013-0137-2 REVIEW PAPER Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering Sirma Yegin & Peter Dekker Received: 25 February 2013 /Revised: 16 May 2013 /Accepted: 21 May 2013 / Published online: 27 June 2013 # INRA and Springer-Verlag France 2013 Abstract Aspartic proteinases are an important class of proteinases which are widely used as milk-coagulating agents in industrial cheese production. They are available from a wide range of sources including mammals, plants, and microorganisms. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Treatment of Cashew Extracts with Aspergillopepsin Reduces Ige Binding to Cashew Allergens
Journal of Applied Biology & Biotechnology Vol. 4 (02), pp. 001-010, March-April, 2016 Available online at http://www.jabonline.in DOI: 10.7324/JABB.2016.40201 Treatment of cashew extracts with Aspergillopepsin reduces IgE binding to cashew allergens Cecily B. DeFreece1, Jeffrey W. Cary2, Casey C. Grimm2, Richard L. Wasserman3, Christopher P Mattison2* 1Department of Biology, Xavier University of Louisiana, New Orleans, LA, USA. 2USDA-ARS, Southern Regional Research Center, New Orleans, LA, USA. 3Allergy Partners of North Texas Research, Department of Pediatrics, Medical City Children’s Hospital, Dallas, Texas, USA. ARTICLE INFO ABSTRACT Article history: Enzymes from Aspergillus fungal species are used in many industrial and pharmaceutical applications. Received on: 21/10/2015 Aspergillus niger and Aspergillus oryzae were cultured on media containing cashew nut flour to identify secreted Revised on: 06/12/2015 proteins that may be useful as future food allergen processing enzymes. Mass-spectrometric analysis of secreted Accepted on: 20/12/2015 proteins and protein bands from SDS-PAGE gels indicated the presence of at least 63 proteins. The majority of Available online: 21/04/2016 these proteins were involved in carbohydrate metabolism, but there were also enzymes involved in lipid and protein metabolism. It is likely that some of these enzymes are specifically upregulated in response to cashew Key words: nut protein, and study of these enzymes could aid our understanding of cashew nut metabolism. Aspergillus, cashew, food Aspergillopepsin from A. niger was one of the proteolytic enzymes identified, and 6 distinct peptides were allergy, immunoglobulin E, matched to this protein providing 22% coverage of the protein. -

Investigating the Impact of Mpapr1, an Aspartic Protease from the Yeast Metschnikowia Pulcherrima, on Wine Properties
THÈSE EN COTUTELLE PRÉSENTÉE POUR OBTENIR LE GRADE DE DOCTEUR DE L’UNIVERSITÉ DE BORDEAUX ET DE L’UNIVERSITÉ DE STELLENBOSCH ÉCOLE DOCTORALE DES SCIENCES DE LA VIE ET DE LA SANTÉ SPÉCIALITÉ ŒNOLOGIE FACULTY OF AGRISCIENCES Par Louwrens THERON ETUDE DE L’IMPACT DE MPAPR1, UNE PROTEASE ASPARTIQUE DE LA LEVURE METSCHNIKOWIA PULCHERRIMA, SUR LES PROPRIETES DU VIN Sous la direction de Benoit DIVOL et de Marina BELY Soutenue le 27 janvier 2017 Membres du jury: Mme. LE HENAFF-LE MARREC Claire, Professeur à l’université de Bordeaux Président M. MARANGON Matteo, Chargé de recherche à l’université de Padoue Rapporteur Mme. CAMARASA Carole, Chargée de recherche à l’INRA de Montpellier Rapporteur M. BAUER Florian, Professeur à l’université de Stellenbosch Examinateur Titre : Etude de l’impact de MpAPr1, une protéase aspartique de la levure Metschnikowia pulcherrima, sur les propriétés du vin Résumé : L'élimination des protéines est une étape clé lors de la production du vin blanc afin d'éviter l'apparition éventuelle d'un voile inoffensif mais inesthétique. Des solutions de rechange à l'utilisation de la bentonite sont activement recherchées en raison des problèmes technologiques, organoleptiques et de durabilité associés à son utilisation. Dans cette étude, MpAPr1, une protéase aspartique extracellulaire préalablement isolée et partiellement caractérisée à partir de la levure Metschnikowia pulcherrima, a été clonée et exprimée de manière hétérologue dans la levure Komagataella pastoris. Les propriétés enzymatiques de MpAPr1 ont été initialement caractérisées dans un extrait brut. Après plusieurs essais faisant appel à différentes techniques, MpAPr1 a été purifié avec succès par chromatographie échangeusede cations. -

(A) Enzymes Derived from Animal Sources
ANNEX (new entries are highlighted in yellow) PERMITTED ENZYMES (A) Enzymes derived from animal sources Enzyme EC Number Source Catalase 1.11.1.6 Bovine liver Lactoperoxidase 1.11.1.7 Bovine milk Bovine stomach; salivary glands or forestomach of calf, kid or lamb; porcine Lipase, triacylglycerol 3.1.1.3 or bovine pancreas Lysozyme 3.2.1.17 Egg whites Pancreatin (or pancreatic elastase) 3.4.21.36 Pancreas of the hog or ox Pepsin 3.4.23.1 Bovine or porcine stomach Phospholipase A2 3.1.1.4 Porcine pancreas Aqueous extracts from the fourth stomach of calves, kids, lambs, and adult Rennet 3.4.23.4 bovine animals, sheep and goats Thrombin 3.4.21.5 Bovine or porcine blood Trypsin 3.4.21.4 Porcine or bovine pancreas (B) Enzymes derived from plant sources Enzyme EC Number Source Alpha–amylase 3.2.1.1 Malted cereals Actinidin 3.4.22.14 Kiwifruit (Actinidia deliciosa) Malted cereals Beta-Amylase 3.2.1.2 Sweet potato (Ipomoea batatas) Bromelain 3.4.22.4 Pineapple fruit/stem (Ananas comosus and Ananas bracteatus (L)) Ficin 3.4.22.3 Ficus spp. Lipoxidase 1.13.11.12 Soyabean whey or meal Papain 3.4.22.2 Carica papaya (L) (Fam. Caricaceae) (C) Enzymes derived from microbial sources EC Enzyme Production organism Donor organism Donor gene Number 1,4-alpha-glucan branching 1,4-alpha-glucan branching 2.4.1.18 Bacillus subtilis Rhodothermus obamensis enzyme enzyme Alpha-acetolactate decarboxylase 4.1.1.5 Bacillus amyloliquefaciens Bacillus subtilis Bacillus subtilis Bacillus brevis Alpha-acetolactate decarboxylase Alpha-amylase 3.2.1.1 Aspergillus niger1 Aspergillus -

Arabidopsis Thaliana Atypical Aspartic Proteases Involved in Primary Root Development and Lateral Root Formation
André Filipe Marques Soares RLR1 and RLR2, two novel Arabidopsis thaliana atypical aspartic proteases involved in primary root development and lateral root formation 2016 Thesis submitted to the Institute for Interdisciplinary Research of the University of Coimbra to apply for the degree of Doctor in Philosophy in the area of Experimental Biology and Biomedicine, specialization in Molecular, Cell and Developmental Biology This work was conducted at the Center for Neuroscience and Cell Biology (CNC) of University of Coimbra and at Biocant - Technology Transfer Association, under the scientific supervision of Doctor Isaura Simões and at the Department of Biochemistry of University of Massachusetts, Amherst, under the scientific supervision of Doctor Alice Y. Cheung. Part of this work was also performed at the Department of Applied Genetics and Cell Biology, University of Natural Resources and Life Sciences, Vienna, under the scientific supervision of Doctor Herta Steinkellner and also at the Central Institute for Engineering, Electronics and Analytics, ZEA-3, Forschungszentrum Jülich, Jülich, under the schientific supervision of Doctor Pitter F. Huesgen. André Filipe Marques Soares was a student of the Doctoral Programme in Experimental Biology and Biomedicine coordinated by the Center for Neuroscience and Cell Biology (CNC) of the University of Coimbra and a recipient of the fellowship SFRH/BD/51676/2011 from the Portuguese Foundation for Science and Technology (FCT). The execution of this work was supported by a PPP grant of the German Academic Exchange Service with funding from the Federal Ministry of Education and Research (Project-ID 57128819 to PFH) and the Fundação para a Ciência e a Tecnologia (FCT) (grant: Scientific and Technological Bilateral Agreement 2015/2016 to IS) Agradecimentos/Acknowledgments Esta tese e todo o percurso que culminou na sua escrita não teriam sido possíveis sem o apoio, o carinho e a amizade de várias pessoas que ainda estão ou estiveram presentes na minha vida. -

Handbook of Proteolytic Enzymes Second Edition Volume 1 Aspartic and Metallo Peptidases
Handbook of Proteolytic Enzymes Second Edition Volume 1 Aspartic and Metallo Peptidases Alan J. Barrett Neil D. Rawlings J. Fred Woessner Editor biographies xxi Contributors xxiii Preface xxxi Introduction ' Abbreviations xxxvii ASPARTIC PEPTIDASES Introduction 1 Aspartic peptidases and their clans 3 2 Catalytic pathway of aspartic peptidases 12 Clan AA Family Al 3 Pepsin A 19 4 Pepsin B 28 5 Chymosin 29 6 Cathepsin E 33 7 Gastricsin 38 8 Cathepsin D 43 9 Napsin A 52 10 Renin 54 11 Mouse submandibular renin 62 12 Memapsin 1 64 13 Memapsin 2 66 14 Plasmepsins 70 15 Plasmepsin II 73 16 Tick heme-binding aspartic proteinase 76 17 Phytepsin 77 18 Nepenthesin 85 19 Saccharopepsin 87 20 Neurosporapepsin 90 21 Acrocylindropepsin 9 1 22 Aspergillopepsin I 92 23 Penicillopepsin 99 24 Endothiapepsin 104 25 Rhizopuspepsin 108 26 Mucorpepsin 11 1 27 Polyporopepsin 113 28 Candidapepsin 115 29 Candiparapsin 120 30 Canditropsin 123 31 Syncephapepsin 125 32 Barrierpepsin 126 33 Yapsin 1 128 34 Yapsin 2 132 35 Yapsin A 133 36 Pregnancy-associated glycoproteins 135 37 Pepsin F 137 38 Rhodotorulapepsin 139 39 Cladosporopepsin 140 40 Pycnoporopepsin 141 Family A2 and others 41 Human immunodeficiency virus 1 retropepsin 144 42 Human immunodeficiency virus 2 retropepsin 154 43 Simian immunodeficiency virus retropepsin 158 44 Equine infectious anemia virus retropepsin 160 45 Rous sarcoma virus retropepsin and avian myeloblastosis virus retropepsin 163 46 Human T-cell leukemia virus type I (HTLV-I) retropepsin 166 47 Bovine leukemia virus retropepsin 169 48 -

EUROPEAN COMMISSION Brussels, 28 April 2020 REGISTER of FOOD
EUROPEAN COMMISSION DIRECTORATE-GENERAL FOR HEALTH AND FOOD SAFETY Food and feed safety, innovation Food processing technologies and novel foods Brussels, 28 April 2020 REGISTER OF FOOD ENZYMES TO BE CONSIDERED FOR INCLUSION IN THE UNION LIST Article 17 of Regulation (EC) No 1332/20081 provides for the establishment of a Register of all food enzymes to be considered for inclusion in the Union list. In accordance with that Article, the Register includes all applications which were submitted within the initial period fixed by that Regulation and which comply with the validity criteria laid down in accordance with Article 9(1) of (EC) No 1331/2008 establishing a common authorisation procedure for food additives, food enzymes and food flavourings2. The Register therefore lists all valid food enzyme applications submitted until 11 March 2015 except those withdrawn by the applicant before that date. Applications submitted after that date are not included in the Register but will be processed in accordance with the Common Authorisation Procedure. The entry of a food enzyme in the Register specifies the identification, the name, the source of the food enzyme as provided by the applicant and the EFSA question number under which the status of the Authority’s assessment can be followed3. As defined by Article 3 of Regulation (EC) No 1332/2008, ‘food enzyme’ subject to an entry in the Register, refers to a product that may contain more than one enzyme capable of catalysing a specific biochemical reaction. In the assessment process, such a food enzyme may be linked with several EFSA question numbers. -

(12) United States Patent (10) Patent No.: US 8,561,811 B2 Bluchel Et Al
USOO8561811 B2 (12) United States Patent (10) Patent No.: US 8,561,811 B2 Bluchel et al. (45) Date of Patent: Oct. 22, 2013 (54) SUBSTRATE FOR IMMOBILIZING (56) References Cited FUNCTIONAL SUBSTANCES AND METHOD FOR PREPARING THE SAME U.S. PATENT DOCUMENTS 3,952,053 A 4, 1976 Brown, Jr. et al. (71) Applicants: Christian Gert Bluchel, Singapore 4.415,663 A 1 1/1983 Symon et al. (SG); Yanmei Wang, Singapore (SG) 4,576,928 A 3, 1986 Tani et al. 4.915,839 A 4, 1990 Marinaccio et al. (72) Inventors: Christian Gert Bluchel, Singapore 6,946,527 B2 9, 2005 Lemke et al. (SG); Yanmei Wang, Singapore (SG) FOREIGN PATENT DOCUMENTS (73) Assignee: Temasek Polytechnic, Singapore (SG) CN 101596422 A 12/2009 JP 2253813 A 10, 1990 (*) Notice: Subject to any disclaimer, the term of this JP 2258006 A 10, 1990 patent is extended or adjusted under 35 WO O2O2585 A2 1, 2002 U.S.C. 154(b) by 0 days. OTHER PUBLICATIONS (21) Appl. No.: 13/837,254 Inaternational Search Report for PCT/SG2011/000069 mailing date (22) Filed: Mar 15, 2013 of Apr. 12, 2011. Suen, Shing-Yi, et al. “Comparison of Ligand Density and Protein (65) Prior Publication Data Adsorption on Dye Affinity Membranes Using Difference Spacer Arms'. Separation Science and Technology, 35:1 (2000), pp. 69-87. US 2013/0210111A1 Aug. 15, 2013 Related U.S. Application Data Primary Examiner — Chester Barry (62) Division of application No. 13/580,055, filed as (74) Attorney, Agent, or Firm — Cantor Colburn LLP application No. -

Purification and Characterization of an Aspartic Protease from The
Electronic Journal of Biotechnology 17 (2014) 89–94 Contents lists available at ScienceDirect Electronic Journal of Biotechnology Purification and characterization of an aspartic protease from the Rhizopus oryzae protease extract, Peptidase R Nai-Wan Hsiao a,1,YehChenb,1,Yi-ChiaKuanb,c, Yen-Chung Lee d, Shuo-Kang Lee b, Hsin-Hua Chan b, Chao-Hung Kao b,⁎ a Institute of Biotechnology, National Changhua University of Education, Changhua 500, Taiwan b Department of Biotechnology, Hungkuang University, Taichung 433, Taiwan c Department of Life Science, National Tsing Hua University, Hsinchu 300, Taiwan d Department of Bioagricultural Science, National Chiayi University, Chiayi 600, Taiwan article info abstract Article history: Background: Aspartic proteases are a subfamily of endopeptidases that are useful in a variety of applications, Received 2 September 2013 especially in the food processing industry. Here we describe a novel aspartic protease that was purified from Accepted 14 January 2014 Peptidase R, a commercial protease preparation derived from Rhizopus oryzae. Available online 17 February 2014 Results: An aspartic protease sourced from Peptidase R was purified to homogeneity by anion exchange chromatography followed by polishing with a hydrophobic interaction chromatography column, resulting in a Keywords: 3.4-fold increase in specificactivity(57.5×103 U/mg) and 58.8% recovery. The estimated molecular weight of Chromatography the purified enzyme was 39 kDa. The N-terminal sequence of the purified protein exhibited 63–75% identity to Endopeptidase – Food processing industry rhizopuspepsins from various Rhizopus species. The enzyme exhibited maximal activity at 75°C in glycine HCl Homogeneity buffer, pH 3.4 with casein as the substrate.