Investigating the Impact of Mpapr1, an Aspartic Protease from the Yeast Metschnikowia Pulcherrima, on Wine Properties
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
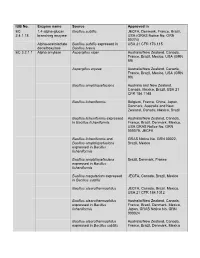
IUB No. Enzyme Name Source Approved in EC 1,4-Alpha-Glucan Bacillus Subtilis JECFA, Denmark, France, Brazil, 2.4.1.18 Branching Enzyme USA (GRAS Notice No
IUB No. Enzyme name Source Approved in EC 1,4-alpha-glucan Bacillus subtilis JECFA, Denmark, France, Brazil, 2.4.1.18 branching enzyme USA (GRAS Notice No. GRN 00274) Alpha-acetolactate Bacillus subtilis expressed in USA 21 CFR 173.115 decarboxylase Bacillus brevis EC 3.2.1.1 Alpha amylase Aspergillus niger Australia/New Zealand, Canada, France, Brazil, Mexico, USA (GRN 89) Aspergillus oryzae Australia/New Zealand, Canada, France, Brazil, Mexico, USA (GRN 90) Bacillus amyloliquefaciens Australia and New Zealand, Canada, Mexico, Brazil, USA 21 CFR 184.1148 Bacillus licheniformis Belgium, France, China, Japan, Denmark, Australia and New Zealand, Canada, Mexico, Brazil Bacillus licheniformis expressed Australia/New Zealand, Canada, in Bacillus licheniformis France, Brazil, Denmark, Mexico, USA GRAS Notice No. GRN 000079, JECFA Bacillus licheniformis and GRAS Notice No. GRN 00022, Bacillus amyloliquefaciens Brazil, Mexico expressed in Bacillus licheniformis Bacillus amyloliquefaciens Brazil, Denmark, France expressed in Bacillus licheniformis Bacillus megaterium expressed JECFA, Canada, Brazil, Mexico in Bacillus subtilis Bacillus stearothermophilus JECFA, Canada, Brazil, Mexico, USA 21 CFR 184.1012 Bacillus stearothermophilus Australia/New Zealand, Canada, expressed in Bacillus France, Brazil, Denmark, Mexico, licheniformis Japan, GRAS Notice No. GRN 000024 Bacillus stearothermophilus Australia/New Zealand, Canada, expressed in Bacillus subtilis France, Brazil, Denmark, Mexico Bacillus stearothermophilus Australia/New Zealand, Canada, (Geobacillus France, Europa, Brazil, Mexico, stearothermophilus) USA (GRN 594) Bacillus subtilis Australia and New Zealand, Canada, Mexico, Brazil, USA 21 CFR 184.1148 Rhizopus delemar Brazil Rhizopus oryzae Brazil, Mexico, USA GRAS Notice No. GRN 000090 Thermoccocales expressed in Brazil, USA (GRN126) Pseudomonas fluorecens Rhizomucor pusillus expressed Brazil, Denmark, France, Mexico, in Aspergillus niger Australia and New Zealand. -

(12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS Et Al
US 2016.0346364A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS et al. (43) Pub. Date: Dec. 1, 2016 (54) MEDICAMENT AND METHOD FOR (52) U.S. Cl. TREATING INNATE IMMUNE RESPONSE CPC ........... A61K 38/488 (2013.01); A61K 38/482 DISEASES (2013.01); C12Y 304/23019 (2013.01); C12Y 304/21026 (2013.01); C12Y 304/23018 (71) Applicant: DSM IPASSETS B.V., Heerlen (NL) (2013.01); A61K 9/0053 (2013.01); C12N 9/62 (2013.01); A23L 29/06 (2016.08); A2ID 8/042 (72) Inventors: Maaike Johanna BRUINS, Kaiseraugst (2013.01); A23L 5/25 (2016.08); A23V (CH); Luppo EDENS, Kaiseraugst 2002/00 (2013.01) (CH); Lenneke NAN, Kaiseraugst (CH) (57) ABSTRACT (21) Appl. No.: 15/101,630 This invention relates to a medicament or a dietary Supple (22) PCT Filed: Dec. 11, 2014 ment comprising the Aspergillus niger aspergilloglutamic peptidase that is capable of hydrolyzing plant food allergens, (86). PCT No.: PCT/EP2014/077355 and more particularly, alpha-amylase/trypsin inhibitors, thereby treating diseases due to an innate immune response S 371 (c)(1), in humans, and/or allowing to delay the onset of said (2) Date: Jun. 3, 2016 diseases. The present invention relates to the discovery that (30) Foreign Application Priority Data the Aspergillus niger aspergilloglutamic peptidase is capable of hydrolyzing alpha-amylase/trypsin inhibitors that are Dec. 11, 2013 (EP) .................................. 13196580.8 present in wheat and related cereals said inhibitors being strong inducers of innate immune response. Furthermore, Publication Classification the present invention relates to a method for hydrolyzing alpha-amylase/trypsin inhibitors comprising incubating a (51) Int. -

Progress in the Field of Aspartic Proteinases in Cheese Manufacturing
Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering Sirma Yegin, Peter Dekker To cite this version: Sirma Yegin, Peter Dekker. Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering. Dairy Science & Technology, EDP sciences/Springer, 2013, 93 (6), pp.565-594. 10.1007/s13594-013-0137-2. hal-01201447 HAL Id: hal-01201447 https://hal.archives-ouvertes.fr/hal-01201447 Submitted on 17 Sep 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Dairy Sci. & Technol. (2013) 93:565–594 DOI 10.1007/s13594-013-0137-2 REVIEW PAPER Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering Sirma Yegin & Peter Dekker Received: 25 February 2013 /Revised: 16 May 2013 /Accepted: 21 May 2013 / Published online: 27 June 2013 # INRA and Springer-Verlag France 2013 Abstract Aspartic proteinases are an important class of proteinases which are widely used as milk-coagulating agents in industrial cheese production. They are available from a wide range of sources including mammals, plants, and microorganisms. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’S Disease Pathology
Review BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’s Disease Pathology Gerald Koelsch CoMentis, Inc., South San Francisco, CA 94080, USA; [email protected] Received: 15 September 2017; Accepted: 10 October 2017; Published: 13 October 2017 Abstract: Alzheimer’s disease (AD) is a fatal progressive neurodegenerative disorder characterized by increasing loss in memory, cognition, and function of daily living. Among the many pathologic events observed in the progression of AD, changes in amyloid β peptide (Aβ) metabolism proceed fastest, and precede clinical symptoms. BACE1 (β-secretase 1) catalyzes the initial cleavage of the amyloid precursor protein to generate Aβ. Therefore inhibition of BACE1 activity could block one of the earliest pathologic events in AD. However, therapeutic BACE1 inhibition to block Aβ production may need to be balanced with possible effects that might result from diminished physiologic functions BACE1, in particular processing of substrates involved in neuronal function of the brain and periphery. Potentials for beneficial or consequential effects resulting from pharmacologic inhibition of BACE1 are reviewed in context of ongoing clinical trials testing the effect of BACE1 candidate inhibitor drugs in AD populations. Keywords: Alzheimer’s disease; amyloid hypothesis; BACE1; beta secretase; pharmacology 1. Introduction Alzheimer’s disease (AD) is a fatal progressive neurodegenerative disorder, slowly eroding memory, cognition, and functions of daily living, inevitably culminating in death from pneumonia and infectious diseases resulting from failure to thrive, loss of fine motor skills, and incapacitation. Treatment is limited to therapeutics that alleviate symptoms of memory loss, but are effective for a relatively short duration during and after which disease progression continues. -

Reduced Reelin Expression Accelerates Amyloid-ßplaque
9228 • The Journal of Neuroscience, July 7, 2010 • 30(27):9228–9240 Neurobiology of Disease Reduced Reelin Expression Accelerates Amyloid- Plaque Formation and Tau Pathology in Transgenic Alzheimer’s Disease Mice Samira Kocherhans,1* Amrita Madhusudan,1* Jana Doehner,1* Karin S. Breu,1 Roger M. Nitsch,2 Jean-Marc Fritschy,1 and Irene Knuesel1 1Institute of Pharmacology and Toxicology, University of Zurich, CH-8057 Zurich, Switzerland, and 2Division of Psychiatry Research, University of Zurich, CH-8008 Zurich, Switzerland In addition to the fundamental role of the extracellular glycoprotein Reelin in neuronal development and adult synaptic plasticity, alterations in Reelin-mediated signaling have been suggested to contribute to neuronal dysfunction associated with Alzheimer’s disease (AD). In vitro data revealed a biochemical link between Reelin-mediated signaling, Tau phosphorylation, and amyloid precursor protein (APP) processing. To directly address the role of Reelin in amyloid- plaque and Tau pathology in vivo, we crossed heterozygous Reelin knock-out mice (reeler) with transgenic AD mice to investigate the temporal and spatial AD-like neuropathology. We demonstrate that a reduction in Reelin expression results in enhanced amyloidogenic APP processing, as indicated by the precocious production of amyloid- peptides, the significant increase in number and size of amyloid- plaques, as well as age-related aggravation of plaque pathology in double mutant compared with single AD mutant mice of both sexes. Numerous amyloid- plaques accumulate in the hippocampal formation and neocortex of double mutants, precisely in layers with strongest Reelin expression and highest accumulation of Reelin plaques in aged wild-type mice. Moreover, concentric accumulations of phosphorylated Tau-positive neurons around amyloid- plaques were evident in 15-month-old double versus single mutant mice. -

Proteolytic Enzymes in Grass Pollen and Their Relationship to Allergenic Proteins
Proteolytic Enzymes in Grass Pollen and their Relationship to Allergenic Proteins By Rohit G. Saldanha A thesis submitted in fulfilment of the requirements for the degree of Masters by Research Faculty of Medicine The University of New South Wales March 2005 TABLE OF CONTENTS TABLE OF CONTENTS 1 LIST OF FIGURES 6 LIST OF TABLES 8 LIST OF TABLES 8 ABBREVIATIONS 8 ACKNOWLEDGEMENTS 11 PUBLISHED WORK FROM THIS THESIS 12 ABSTRACT 13 1. ASTHMA AND SENSITISATION IN ALLERGIC DISEASES 14 1.1 Defining Asthma and its Clinical Presentation 14 1.2 Inflammatory Responses in Asthma 15 1.2.1 The Early Phase Response 15 1.2.2 The Late Phase Reaction 16 1.3 Effects of Airway Inflammation 16 1.3.1 Respiratory Epithelium 16 1.3.2 Airway Remodelling 17 1.4 Classification of Asthma 18 1.4.1 Extrinsic Asthma 19 1.4.2 Intrinsic Asthma 19 1.5 Prevalence of Asthma 20 1.6 Immunological Sensitisation 22 1.7 Antigen Presentation and development of T cell Responses. 22 1.8 Factors Influencing T cell Activation Responses 25 1.8.1 Co-Stimulatory Interactions 25 1.8.2 Cognate Cellular Interactions 26 1.8.3 Soluble Pro-inflammatory Factors 26 1.9 Intracellular Signalling Mechanisms Regulating T cell Differentiation 30 2 POLLEN ALLERGENS AND THEIR RELATIONSHIP TO PROTEOLYTIC ENZYMES 33 1 2.1 The Role of Pollen Allergens in Asthma 33 2.2 Environmental Factors influencing Pollen Exposure 33 2.3 Classification of Pollen Sources 35 2.3.1 Taxonomy of Pollen Sources 35 2.3.2 Cross-Reactivity between different Pollen Allergens 40 2.4 Classification of Pollen Allergens 41 2.4.1 -

Review Article the Role of Microbial Aspartic Protease Enzyme in Food and Beverage Industries
Hindawi Journal of Food Quality Volume 2018, Article ID 7957269, 15 pages https://doi.org/10.1155/2018/7957269 Review Article The Role of Microbial Aspartic Protease Enzyme in Food and Beverage Industries Jermen Mamo and Fassil Assefa Microbial, Cellular and Molecular Biology Department, College of Natural Science, Addis Ababa University, P.O. Box 1176, Addis Ababa, Ethiopia Correspondence should be addressed to Jermen Mamo; [email protected] Received 3 April 2018; Revised 16 May 2018; Accepted 29 May 2018; Published 3 July 2018 Academic Editor: Antimo Di Maro Copyright © 2018 Jermen Mamo and Fassil Assefa. is is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Proteases represent one of the three largest groups of industrial enzymes and account for about 60% of the total global enzymes sale. According to the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology, proteases are classied in enzymes of class 3, the hydrolases, and the subclass 3.4, the peptide hydrolases or peptidase. Proteases are generally grouped into two main classes based on their site of action, that is, exopeptidases and endopeptidases. Protease has also been grouped into four classes based on their catalytic action: aspartic, cysteine, metallo, and serine proteases. However, lately, three new systems have been dened: the threonine-based proteasome system, the glutamate-glutamine system of eqolisin, and the serine-glutamate-aspartate system of sedolisin. Aspartic proteases (EC 3.4.23) are peptidases that display various activities and specicities. -

Treatment of Cashew Extracts with Aspergillopepsin Reduces Ige Binding to Cashew Allergens
Journal of Applied Biology & Biotechnology Vol. 4 (02), pp. 001-010, March-April, 2016 Available online at http://www.jabonline.in DOI: 10.7324/JABB.2016.40201 Treatment of cashew extracts with Aspergillopepsin reduces IgE binding to cashew allergens Cecily B. DeFreece1, Jeffrey W. Cary2, Casey C. Grimm2, Richard L. Wasserman3, Christopher P Mattison2* 1Department of Biology, Xavier University of Louisiana, New Orleans, LA, USA. 2USDA-ARS, Southern Regional Research Center, New Orleans, LA, USA. 3Allergy Partners of North Texas Research, Department of Pediatrics, Medical City Children’s Hospital, Dallas, Texas, USA. ARTICLE INFO ABSTRACT Article history: Enzymes from Aspergillus fungal species are used in many industrial and pharmaceutical applications. Received on: 21/10/2015 Aspergillus niger and Aspergillus oryzae were cultured on media containing cashew nut flour to identify secreted Revised on: 06/12/2015 proteins that may be useful as future food allergen processing enzymes. Mass-spectrometric analysis of secreted Accepted on: 20/12/2015 proteins and protein bands from SDS-PAGE gels indicated the presence of at least 63 proteins. The majority of Available online: 21/04/2016 these proteins were involved in carbohydrate metabolism, but there were also enzymes involved in lipid and protein metabolism. It is likely that some of these enzymes are specifically upregulated in response to cashew Key words: nut protein, and study of these enzymes could aid our understanding of cashew nut metabolism. Aspergillus, cashew, food Aspergillopepsin from A. niger was one of the proteolytic enzymes identified, and 6 distinct peptides were allergy, immunoglobulin E, matched to this protein providing 22% coverage of the protein. -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Protein secretion and encystation in Acanthamoeba Alvaro de Obeso Fernández del Valle Doctor of Philosophy The University of Edinburgh 2018 Abstract Free-living amoebae (FLA) are protists of ubiquitous distribution characterised by their changing morphology and their crawling movements. They have no common phylogenetic origin but can be found in most protist evolutionary branches. Acanthamoeba is a common FLA that can be found worldwide and is capable of infecting humans. The main disease is a life altering infection of the cornea named Acanthamoeba keratitis. Additionally, Acanthamoeba has a close relationship to bacteria. Acanthamoeba feeds on bacteria. At the same time, some bacteria have adapted to survive inside Acanthamoeba and use it as transport or protection to increase survival. When conditions are adverse, Acanthamoeba is capable of differentiating into a protective cyst. -

(A) Enzymes Derived from Animal Sources
ANNEX (new entries are highlighted in yellow) PERMITTED ENZYMES (A) Enzymes derived from animal sources Enzyme EC Number Source Catalase 1.11.1.6 Bovine liver Lactoperoxidase 1.11.1.7 Bovine milk Bovine stomach; salivary glands or forestomach of calf, kid or lamb; porcine Lipase, triacylglycerol 3.1.1.3 or bovine pancreas Lysozyme 3.2.1.17 Egg whites Pancreatin (or pancreatic elastase) 3.4.21.36 Pancreas of the hog or ox Pepsin 3.4.23.1 Bovine or porcine stomach Phospholipase A2 3.1.1.4 Porcine pancreas Aqueous extracts from the fourth stomach of calves, kids, lambs, and adult Rennet 3.4.23.4 bovine animals, sheep and goats Thrombin 3.4.21.5 Bovine or porcine blood Trypsin 3.4.21.4 Porcine or bovine pancreas (B) Enzymes derived from plant sources Enzyme EC Number Source Alpha–amylase 3.2.1.1 Malted cereals Actinidin 3.4.22.14 Kiwifruit (Actinidia deliciosa) Malted cereals Beta-Amylase 3.2.1.2 Sweet potato (Ipomoea batatas) Bromelain 3.4.22.4 Pineapple fruit/stem (Ananas comosus and Ananas bracteatus (L)) Ficin 3.4.22.3 Ficus spp. Lipoxidase 1.13.11.12 Soyabean whey or meal Papain 3.4.22.2 Carica papaya (L) (Fam. Caricaceae) (C) Enzymes derived from microbial sources EC Enzyme Production organism Donor organism Donor gene Number 1,4-alpha-glucan branching 1,4-alpha-glucan branching 2.4.1.18 Bacillus subtilis Rhodothermus obamensis enzyme enzyme Alpha-acetolactate decarboxylase 4.1.1.5 Bacillus amyloliquefaciens Bacillus subtilis Bacillus subtilis Bacillus brevis Alpha-acetolactate decarboxylase Alpha-amylase 3.2.1.1 Aspergillus niger1 Aspergillus -

Molecular Sciences a New Pepstatin-Insensitive Thermopsin
Int. J. Mol. Sci. 2014, 15, 3204-3219; doi:10.3390/ijms15023204 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article A New Pepstatin-Insensitive Thermopsin-Like Protease Overproduced in Peptide-Rich Cultures of Sulfolobus solfataricus Marta Gogliettino †, Alessia Riccio †, Ennio Cocca, Mosè Rossi, Gianna Palmieri * and Marco Balestrieri Institute of Biosciences and BioResources, National Research Council (CNR-IBBR), Via Pietro Castellino 111, Naples 80131, Italy; E-Mails: [email protected] (M.G.); [email protected] (A.R.); [email protected] (E.C.); [email protected] (M.R.); [email protected] (M.B.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +39-081-6132-711; Fax: +39-081-6132-277. Received: 5 December 2013; in revised form: 26 January 2014 / Accepted: 11 February 2014 / Published: 21 February 2014 Abstract: In this study, we gain insight into the extracellular proteolytic system of Sulfolobus solfataricus grown on proteinaceous substrates, providing further evidence that acidic proteases were specifically produced in response to peptide-rich media. The main proteolytic component was the previously isolated SsMTP (Sulfolobus solfataricus multi-domain thermopsin-like protease), while the less abundant (named SsMTP-1) one was purified, characterized and identified as the sso1175 gene-product. The protein revealed a multi-domain organization shared with the cognate SsMTP with a catalytic domain followed by several tandemly-repeated motifs. Moreover, both enzymes were found spread across the Crenarchaeota phylum and belonging to the thermopsin family, although segregated into diverse phylogenetic clusters.