Arabidopsis Thaliana Atypical Aspartic Proteases Involved in Primary Root Development and Lateral Root Formation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
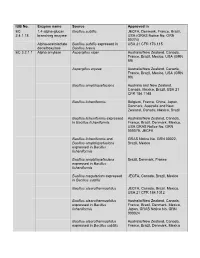
IUB No. Enzyme Name Source Approved in EC 1,4-Alpha-Glucan Bacillus Subtilis JECFA, Denmark, France, Brazil, 2.4.1.18 Branching Enzyme USA (GRAS Notice No
IUB No. Enzyme name Source Approved in EC 1,4-alpha-glucan Bacillus subtilis JECFA, Denmark, France, Brazil, 2.4.1.18 branching enzyme USA (GRAS Notice No. GRN 00274) Alpha-acetolactate Bacillus subtilis expressed in USA 21 CFR 173.115 decarboxylase Bacillus brevis EC 3.2.1.1 Alpha amylase Aspergillus niger Australia/New Zealand, Canada, France, Brazil, Mexico, USA (GRN 89) Aspergillus oryzae Australia/New Zealand, Canada, France, Brazil, Mexico, USA (GRN 90) Bacillus amyloliquefaciens Australia and New Zealand, Canada, Mexico, Brazil, USA 21 CFR 184.1148 Bacillus licheniformis Belgium, France, China, Japan, Denmark, Australia and New Zealand, Canada, Mexico, Brazil Bacillus licheniformis expressed Australia/New Zealand, Canada, in Bacillus licheniformis France, Brazil, Denmark, Mexico, USA GRAS Notice No. GRN 000079, JECFA Bacillus licheniformis and GRAS Notice No. GRN 00022, Bacillus amyloliquefaciens Brazil, Mexico expressed in Bacillus licheniformis Bacillus amyloliquefaciens Brazil, Denmark, France expressed in Bacillus licheniformis Bacillus megaterium expressed JECFA, Canada, Brazil, Mexico in Bacillus subtilis Bacillus stearothermophilus JECFA, Canada, Brazil, Mexico, USA 21 CFR 184.1012 Bacillus stearothermophilus Australia/New Zealand, Canada, expressed in Bacillus France, Brazil, Denmark, Mexico, licheniformis Japan, GRAS Notice No. GRN 000024 Bacillus stearothermophilus Australia/New Zealand, Canada, expressed in Bacillus subtilis France, Brazil, Denmark, Mexico Bacillus stearothermophilus Australia/New Zealand, Canada, (Geobacillus France, Europa, Brazil, Mexico, stearothermophilus) USA (GRN 594) Bacillus subtilis Australia and New Zealand, Canada, Mexico, Brazil, USA 21 CFR 184.1148 Rhizopus delemar Brazil Rhizopus oryzae Brazil, Mexico, USA GRAS Notice No. GRN 000090 Thermoccocales expressed in Brazil, USA (GRN126) Pseudomonas fluorecens Rhizomucor pusillus expressed Brazil, Denmark, France, Mexico, in Aspergillus niger Australia and New Zealand. -

(12) United States Patent (10) Patent No.: US 6,395,889 B1 Robison (45) Date of Patent: May 28, 2002
USOO6395889B1 (12) United States Patent (10) Patent No.: US 6,395,889 B1 Robison (45) Date of Patent: May 28, 2002 (54) NUCLEIC ACID MOLECULES ENCODING WO WO-98/56804 A1 * 12/1998 ........... CO7H/21/02 HUMAN PROTEASE HOMOLOGS WO WO-99/0785.0 A1 * 2/1999 ... C12N/15/12 WO WO-99/37660 A1 * 7/1999 ........... CO7H/21/04 (75) Inventor: fish E. Robison, Wilmington, MA OTHER PUBLICATIONS Vazquez, F., et al., 1999, “METH-1, a human ortholog of (73) Assignee: Millennium Pharmaceuticals, Inc., ADAMTS-1, and METH-2 are members of a new family of Cambridge, MA (US) proteins with angio-inhibitory activity', The Journal of c: - 0 Biological Chemistry, vol. 274, No. 33, pp. 23349–23357.* (*) Notice: Subject to any disclaimer, the term of this Descriptors of Protease Classes in Prosite and Pfam Data patent is extended or adjusted under 35 bases. U.S.C. 154(b) by 0 days. * cited by examiner (21) Appl. No.: 09/392, 184 Primary Examiner Ponnathapu Achutamurthy (22) Filed: Sep. 9, 1999 ASSistant Examiner William W. Moore (51) Int. Cl." C12N 15/57; C12N 15/12; (74) Attorney, Agent, or Firm-Alston & Bird LLP C12N 9/64; C12N 15/79 (57) ABSTRACT (52) U.S. Cl. .................... 536/23.2; 536/23.5; 435/69.1; 435/252.3; 435/320.1 The invention relates to polynucleotides encoding newly (58) Field of Search ............................... 536,232,235. identified protease homologs. The invention also relates to 435/6, 226, 69.1, 252.3 the proteases. The invention further relates to methods using s s s/ - - -us the protease polypeptides and polynucleotides as a target for (56) References Cited diagnosis and treatment in protease-mediated disorders. -

In Vitro Inhibition of HIV-1 Proteinase by Cerulenin
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 261, number 2, 373-377 FEBS 08165 February 1990 In vitro inhibition of HIV-1 proteinase by cerulenin Karin Moelling, Thomas Schulze, Marie-Theres Knoop, John Kay +, Raymond Jupp +, George Nicolaou* and Laurence H. Pearl* Max-Planck lnstitut fiir Molekular Genetik, lhnestrasse 73, D-IO00 Berlin 33, FRG, +Department of Biochemistry, University College of Wales, PO Box 903, Cardiff CFl 1ST, UK and *Department of Biochemistry, University College London, Gower Street, London WCIE 6BT, UK Received 13 October 1989; revised version received 18 December 1989 Retroviruses encode proteinases necessary for the proteolytic processing of the viral gag and gag-pol precursor proteins. These enzymes have been shown to be structurally and functionally related to aspartyl proteinases such as pepsin and renin. Cerulenin is a naturally occurring antibiotic, commonly used as an inhibitor of fatty acid synthesis. Cerulenin has been observed to inhibit production of Rous sarcoma virus and murine leukae- mia virus by infected cells, possibly by interfering with proteolytic processing of viral precursor proteins. We show here that cerulenin inhibits the action of the HIV-1 proteinase in vitro, using 3 substrates: a synthetic heptapeptide (SQNYPIV) which corresponds to the sequence at the HIV-I gag p17/p24 junction, a bacterially expressed gag precursor, and purified 66 kDa reverse transcriptase. Inhibition of cleavage by HIV-1 proteinase required preincubation with cerulenin. Cerulenin also inactivates endothiapepsin, a well-characterised fungal aspartyl proteinase, sug- gesting that the action of cerulenin is a function of the common active site structure of the retroviral and aspartic proteinases. -

Chymotrypsin-Like Peptidases in Insects
Chymotrypsin-like peptidases in insects Dissertation zur Erlangung des akademischen Grades Doctor rerum naturalium (Dr. rer. nat.) Fachbereich Biologie/Chemie der Universität Osnabrück vorgelegt von Gunnar Bröhan Osnabrück, Mai 2010 TABLE OF CONTENTS I Table of contents 1. Introduction 1 1.1. Serine endopeptidases 1 1.2. The structure of S1A chymotrypsin-like peptidases 2 1.3. Catalytic mechanism of chymotrypsin-like peptidases 6 1.4. Insect chymotrypsin-like peptidases 9 1.4.1. Chymotrypsin-like peptidases in insect immunity 9 1.4.2. Role of chymotrypsin-like peptidases in digestion 14 1.4.3. Involvement of chymotrypsin-like peptidases in molt 16 1.5. Objective of the work 18 2. Material and Methods 20 2.1. Material 20 2.1.1. Culture Media 20 2.1.2. Insects 20 2.2. Molecular biological methods 20 2.2.1. Tissue preparations for total RNA isolation 20 2.2.2. Total RNA isolation 21 2.2.3. Reverse transcription 21 2.2.4. Quantification of nucleic acids 21 2.2.5. Chemical competent Escherichia coli 21 2.2.6. Ligation and transformation in E. coli 21 2.2.7. Preparation of plasmid DNA 22 2.2.8. Restriction enzyme digestion of DNA 22 2.2.9. DNA gel-electrophoresis and DNA isolation 22 2.2.10. Polymerase-chain-reaction based methods 23 2.2.10.1. RACE-PCR 23 2.2.10.2. Quantitative Realtime PCR 23 2.2.10.3. Megaprimer PCR 24 2.2.11. Cloning of insect CTLPs 25 2.2.12. Syntheses of Digoxigenin-labeled DNA and RNA probes 26 2.2.13. -

Concentration of Tissue Angiotensin II Increases with Severity of Experimental Pancreatitis
MOLECULAR MEDICINE REPORTS 8: 335-338, 2013 Concentration of tissue angiotensin II increases with severity of experimental pancreatitis HIROYUKI FURUKAWA1, ATSUSHI SHINMURA1, HIDEHIRO TAJIMA1, TOMOYA TSUKADA1, SHIN-ICHI NAKANUMA1, KOICHI OKAMOTO1, SEISHO SAKAI1, ISAMU MAKINO1, KEISHI NAKAMURA1, HIRONORI HAYASHI1, KATSUNOBU OYAMA1, MASAFUMI INOKUCHI1, HISATOSHI NAKAGAWARA1, TOMOHARU MIYASHITA1, HIDETO FUJITA1, HIROYUKI TAKAMURA1, ITASU NINOMIYA1; HIROHISA KITAGAWA1, SACHIO FUSHIDA1, TAKASHI FUJIMURA1, TETSUO OHTA1, TOMOHIKO WAKAYAMA2 and SHOICHI ISEKI2 1Department of Gastroenterological Surgery, Division of Cancer Medicine; 2Department of Histology and Embryology, Graduate School of Medical Science, Kanazawa University, Kanazawa, Ishikawa 920-8641, Japan Received January 23, 2013; Accepted May 30, 2013 DOI: 10.3892/mmr.2013.1509 Abstract. Necrotizing pancreatitis is a serious condition that Introduction is associated with high morbidity and mortality. Although vasospasm is reportedly involved in necrotizing pancreatitis, Acute pancreatitis, particularly the necrotizing type, is the underlying mechanism is not completely clear. In addition, associated with high morbidity and mortality. Necrotizing the local renin-angiotensin system has been hypothesized to pancreatitis is characterized by parenchymal non-enhance- be involved in the progression of pancreatitis and trypsin has ment on contrast-enhanced computed tomography images (1). been shown to generate angiotensin II under weakly acidic Although necrosis is irreversible, certain patients exhibiting conditions. However, to the best of our knowledge, no studies this pancreatic parenchymal non-enhancement recover have reported elevated angiotensin II levels in tissue with with normal pancreatic conditions (2). Vasospasm has been pancreatitis. In the present study, the concentration of pancre- implicated in the development of pancreatic ischemia and atic angiotensin II in rats with experimentally induced acute necrosis (3); however, the precise underlying mechanism is pancreatitis was measured. -

In Escherichia Coli (Synthetic Oligonucleotide/Gene Expression/Industrial Enzyme) J
Proc. Nati Acad. Sci. USA Vol. 80, pp. 3671-3675, June 1983 Biochemistry Synthesis of calf prochymosin (prorennin) in Escherichia coli (synthetic oligonucleotide/gene expression/industrial enzyme) J. S. EMTAGE*, S. ANGALt, M. T. DOELt, T. J. R. HARRISt, B. JENKINS*, G. LILLEYt, AND P. A. LOWEt Departments of *Molecular Genetics, tMolecular Biology, and tFermentation Development, Celltech Limited, 250 Bath Road, Slough SL1 4DY, Berkshire, United Kingdom Communicated by Sydney Brenner, March 23, 1983 ABSTRACT A gene for calf prochymosin (prorennin) has been maturation conditions and remained insoluble on neutraliza- reconstructed from chemically synthesized oligodeoxyribonucleo- tion, it was possible that purification as well as activation could tides and cloned DNA copies of preprochymosin mRNA. This gene be achieved. has been inserted into a bacterial expression plasmid containing We describe here the construction of E. coli plasmids de- the Escherichia coli tryptophan promoter and a bacterial ribo- signed to express the prochymosin gene from the trp promoter some binding site. Induction oftranscription from the tryptophan and the isolation and conversion of this prochymosin to en- promoter results in prochymosin synthesis at a level of up to 5% active of total protein. The enzyme has been purified from bacteria by zymatically chymosin. extraction with urea and chromatography on DEAE-celiulose and MATERIALS AND METHODS converted to enzymatically active chymosin by acidification and neutralization. Bacterially produced chymosin is as effective in Materials. DNase I, pepstatin A, and phenylmethylsulfonyl clotting milk as the natural enzyme isolated from calf stomach. fluoride were obtained from Sigma. Calf prochymosin (Mr 40,431) and chymosin (Mr 35,612) were purified from stomachs Chymosin (rennin) is an aspartyl proteinase found in the fourth from 1-day-old calves (1). -

Membrane Proteins • Cofactors – Plimstex • Membranes • Dna • Small Molecules/Gas • Large Complexes
Structural mass spectrometry hydrogen/deuterium exchange Petr Man Structural Biology and Cell Signalling Institute of Microbiology, Czech Academy of Sciences Structural biology methods Low-resolution methods High-resolution methods Rigid SAXS IR Raman CD ITC MST Cryo-EM AUC SPR MS X-ray crystallography Chemical cross-linking H/D exchange Native ESI + ion mobility Oxidative labelling Small Large NMR Dynamic Structural biology approaches Simple MS, quantitative MS Cross-linking, top-down, native MS+dissociation native MS+ion mobility Cross-linking Structural MS What can we get using mass spectrometry IM – ion mobility CXL – chemical cross-linking AP – afinity purification OFP – oxidative footprinting HDX – hydrogen/deuterium exchange ISOTOPE EXCHANGE IN PROTEINS 1H 2H 3H occurence [%] 99.988 0.0115 trace 5 …Kaj Ulrik Linderstrøm-Lang „Cartesian diver“ Proteins are migrating in tubes with density gradient until they stop at the point where the densities are equal 1H 2H 3H % 99.9885 0.0115 trace density [g/cm3] 1.000 1.106 1.215 Methods of detection IR: β-: NMR: 1 n = 1.6749 × 10-27 kg MS: 1H 2H 3H výskyt% [%] 99.9885 0.0115 trace hustotadensity vody [g/cm [g/cm3] 3] 1.000 1.106 1.215 jadernýspinspin ½+ 1+ ½+ mass [u] 1.00783 2.01410 3.01605 Factors affecting H/D exchange hydrogen bonding solvent accessibility Factors affecting H/D exchange Side chains (acidity, steric shielding) Bai et al.: Proteins (1993) Glasoe, Long: J. Phys. Chem. (1960) Factors affecting H/D exchange – side chain effects Inductive effect – electron density is Downward shift due to withdrawn from peptide steric hindrance effect of bond (S, O). -

(12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS Et Al
US 2016.0346364A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS et al. (43) Pub. Date: Dec. 1, 2016 (54) MEDICAMENT AND METHOD FOR (52) U.S. Cl. TREATING INNATE IMMUNE RESPONSE CPC ........... A61K 38/488 (2013.01); A61K 38/482 DISEASES (2013.01); C12Y 304/23019 (2013.01); C12Y 304/21026 (2013.01); C12Y 304/23018 (71) Applicant: DSM IPASSETS B.V., Heerlen (NL) (2013.01); A61K 9/0053 (2013.01); C12N 9/62 (2013.01); A23L 29/06 (2016.08); A2ID 8/042 (72) Inventors: Maaike Johanna BRUINS, Kaiseraugst (2013.01); A23L 5/25 (2016.08); A23V (CH); Luppo EDENS, Kaiseraugst 2002/00 (2013.01) (CH); Lenneke NAN, Kaiseraugst (CH) (57) ABSTRACT (21) Appl. No.: 15/101,630 This invention relates to a medicament or a dietary Supple (22) PCT Filed: Dec. 11, 2014 ment comprising the Aspergillus niger aspergilloglutamic peptidase that is capable of hydrolyzing plant food allergens, (86). PCT No.: PCT/EP2014/077355 and more particularly, alpha-amylase/trypsin inhibitors, thereby treating diseases due to an innate immune response S 371 (c)(1), in humans, and/or allowing to delay the onset of said (2) Date: Jun. 3, 2016 diseases. The present invention relates to the discovery that (30) Foreign Application Priority Data the Aspergillus niger aspergilloglutamic peptidase is capable of hydrolyzing alpha-amylase/trypsin inhibitors that are Dec. 11, 2013 (EP) .................................. 13196580.8 present in wheat and related cereals said inhibitors being strong inducers of innate immune response. Furthermore, Publication Classification the present invention relates to a method for hydrolyzing alpha-amylase/trypsin inhibitors comprising incubating a (51) Int. -

Structure-Based Optimization of Inhibitors of the Aspartic Protease Endothiapepsin
Int. J. Mol. Sci. 2015, 16, 19184-19194; doi:10.3390/ijms160819184 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Structure-Based Optimization of Inhibitors of the Aspartic Protease Endothiapepsin Alwin M. Hartman 1,†, Milon Mondal 1,†, Nedyalka Radeva 2, Gerhard Klebe 2 and Anna K. H. Hirsch 1,* 1 Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands; E-Mails: [email protected] (A.M.H.); [email protected] (M.M.) 2 Institute of Pharmaceutical Chemistry, Philipps-University Marburg, Marbacher Weg 6, 35032 Marburg, Germany; E-Mails: [email protected] (N.R.); [email protected] (G.K.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Fax: +31-50-363-4296. Academic Editor: John George Hardy Received: 1 May 2015 / Accepted: 6 July 2015 / Published: 14 August 2015 Abstract: Aspartic proteases are a class of enzymes that play a causative role in numerous diseases such as malaria (plasmepsins), Alzheimer’s disease (β-secretase), fungal infections (secreted aspartic proteases), and hypertension (renin). We have chosen endothiapepsin as a model enzyme of this class of enzymes, for the design, preparation and biochemical evaluation of a new series of inhibitors of endothiapepsin. Here, we have optimized a hit, identified by de novo structure-based drug design (SBDD) and DCC, by using structure-based design approaches focusing on the optimization of an amide–π interaction. Biochemical results are in agreement with SBDD. -

Progress in the Field of Aspartic Proteinases in Cheese Manufacturing
Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering Sirma Yegin, Peter Dekker To cite this version: Sirma Yegin, Peter Dekker. Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering. Dairy Science & Technology, EDP sciences/Springer, 2013, 93 (6), pp.565-594. 10.1007/s13594-013-0137-2. hal-01201447 HAL Id: hal-01201447 https://hal.archives-ouvertes.fr/hal-01201447 Submitted on 17 Sep 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Dairy Sci. & Technol. (2013) 93:565–594 DOI 10.1007/s13594-013-0137-2 REVIEW PAPER Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering Sirma Yegin & Peter Dekker Received: 25 February 2013 /Revised: 16 May 2013 /Accepted: 21 May 2013 / Published online: 27 June 2013 # INRA and Springer-Verlag France 2013 Abstract Aspartic proteinases are an important class of proteinases which are widely used as milk-coagulating agents in industrial cheese production. They are available from a wide range of sources including mammals, plants, and microorganisms. -

Discovery of Digestive Enzymes in Carnivorous Plants with Focus on Proteases
A peer-reviewed version of this preprint was published in PeerJ on 5 June 2018. View the peer-reviewed version (peerj.com/articles/4914), which is the preferred citable publication unless you specifically need to cite this preprint. Ravee R, Mohd Salleh F‘, Goh H. 2018. Discovery of digestive enzymes in carnivorous plants with focus on proteases. PeerJ 6:e4914 https://doi.org/10.7717/peerj.4914 Discovery of digestive enzymes in carnivorous plants with focus on proteases Rishiesvari Ravee 1 , Faris ‘Imadi Mohd Salleh 1 , Hoe-Han Goh Corresp. 1 1 Institute of Systems Biology (INBIOSIS), Universiti Kebangsaan Malaysia, Bangi, Selangor, Malaysia Corresponding Author: Hoe-Han Goh Email address: [email protected] Background. Carnivorous plants have been fascinating researchers with their unique characters and bioinspired applications. These include medicinal trait of some carnivorous plants with potentials for pharmaceutical industry. Methods. This review will cover recent progress based on current studies on digestive enzymes secreted by different genera of carnivorous plants: Drosera (sundews), Dionaea (Venus flytrap), Nepenthes (tropical pitcher plants), Sarracenia (North American pitcher plants), Cephalotus (Australian pitcher plants), Genlisea (corkscrew plants), and Utricularia (bladderworts). Results. Since the discovery of secreted protease nepenthesin in Nepenthes pitcher, digestive enzymes from carnivorous plants have been the focus of many studies. Recent genomics approaches have accelerated digestive enzyme discovery. Furthermore, the advancement in recombinant technology and protein purification helped in the identification and characterisation of enzymes in carnivorous plants. Discussion. These different aspects will be described and discussed in this review with focus on the role of secreted plant proteases and their potential industrial applications. -
![A Label-Free Cellular Proteomics Approach to Decipher the Antifungal Action of Dimiq, a Potent Indolo[2,3- B]Quinoline Agent, Against Candida Albicans Biofilms](https://docslib.b-cdn.net/cover/9827/a-label-free-cellular-proteomics-approach-to-decipher-the-antifungal-action-of-dimiq-a-potent-indolo-2-3-b-quinoline-agent-against-candida-albicans-biofilms-409827.webp)
A Label-Free Cellular Proteomics Approach to Decipher the Antifungal Action of Dimiq, a Potent Indolo[2,3- B]Quinoline Agent, Against Candida Albicans Biofilms
A Label-Free Cellular Proteomics Approach to Decipher the Antifungal Action of DiMIQ, a Potent Indolo[2,3- b]Quinoline Agent, against Candida albicans Biofilms Robert Zarnowski 1,2*, Anna Jaromin 3*, Agnieszka Zagórska 4, Eddie G. Dominguez 1,2, Katarzyna Sidoryk 5, Jerzy Gubernator 3 and David R. Andes 1,2 1 Department of Medicine, School of Medicine & Public Health, University of Wisconsin-Madison, Madison, WI 53706, USA; [email protected] (E.G.D.); [email protected] (D.R.A.) 2 Department of Medical Microbiology, School of Medicine & Public Health, University of Wisconsin-Madison, Madison, WI 53706, USA 3 Department of Lipids and Liposomes, Faculty of Biotechnology, University of Wroclaw, 50-383 Wroclaw, Poland; [email protected] 4 Department of Medicinal Chemistry, Jagiellonian University Medical College, 30-688 Cracow, Poland; [email protected] 5 Department of Pharmacy, Cosmetic Chemicals and Biotechnology, Team of Chemistry, Łukasiewicz Research Network-Industrial Chemistry Institute, 01-793 Warsaw, Poland; [email protected] * Correspondence: [email protected] (R.Z.); [email protected] (A.J.); Tel.: +1-608-265-8578 (R.Z.); +48-71-3756203 (A.J.) Label-Free Cellular Proteomics of Candida albicans biofilms treated with DiMIQ Identified Proteins Accession # Alternate ID Gene names (ORF ) WT DIMIQ Z SCORE Proteins induced by DiMIQ Arginase (EC 3.5.3.1) A0A1D8PP00 CAR1 CAALFM_C504490CA 0.000 6.648 drug induced Glucan 1,3-beta-glucosidase BGL2 (EC 3.2.1.58) (Exo-1Q5AMT2 BGL2 CAALFM_C402250CA