Purification and Characterization of an Aspartic Protease from The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
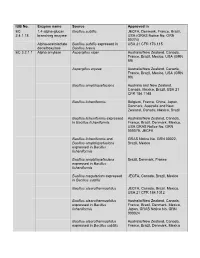
IUB No. Enzyme Name Source Approved in EC 1,4-Alpha-Glucan Bacillus Subtilis JECFA, Denmark, France, Brazil, 2.4.1.18 Branching Enzyme USA (GRAS Notice No
IUB No. Enzyme name Source Approved in EC 1,4-alpha-glucan Bacillus subtilis JECFA, Denmark, France, Brazil, 2.4.1.18 branching enzyme USA (GRAS Notice No. GRN 00274) Alpha-acetolactate Bacillus subtilis expressed in USA 21 CFR 173.115 decarboxylase Bacillus brevis EC 3.2.1.1 Alpha amylase Aspergillus niger Australia/New Zealand, Canada, France, Brazil, Mexico, USA (GRN 89) Aspergillus oryzae Australia/New Zealand, Canada, France, Brazil, Mexico, USA (GRN 90) Bacillus amyloliquefaciens Australia and New Zealand, Canada, Mexico, Brazil, USA 21 CFR 184.1148 Bacillus licheniformis Belgium, France, China, Japan, Denmark, Australia and New Zealand, Canada, Mexico, Brazil Bacillus licheniformis expressed Australia/New Zealand, Canada, in Bacillus licheniformis France, Brazil, Denmark, Mexico, USA GRAS Notice No. GRN 000079, JECFA Bacillus licheniformis and GRAS Notice No. GRN 00022, Bacillus amyloliquefaciens Brazil, Mexico expressed in Bacillus licheniformis Bacillus amyloliquefaciens Brazil, Denmark, France expressed in Bacillus licheniformis Bacillus megaterium expressed JECFA, Canada, Brazil, Mexico in Bacillus subtilis Bacillus stearothermophilus JECFA, Canada, Brazil, Mexico, USA 21 CFR 184.1012 Bacillus stearothermophilus Australia/New Zealand, Canada, expressed in Bacillus France, Brazil, Denmark, Mexico, licheniformis Japan, GRAS Notice No. GRN 000024 Bacillus stearothermophilus Australia/New Zealand, Canada, expressed in Bacillus subtilis France, Brazil, Denmark, Mexico Bacillus stearothermophilus Australia/New Zealand, Canada, (Geobacillus France, Europa, Brazil, Mexico, stearothermophilus) USA (GRN 594) Bacillus subtilis Australia and New Zealand, Canada, Mexico, Brazil, USA 21 CFR 184.1148 Rhizopus delemar Brazil Rhizopus oryzae Brazil, Mexico, USA GRAS Notice No. GRN 000090 Thermoccocales expressed in Brazil, USA (GRN126) Pseudomonas fluorecens Rhizomucor pusillus expressed Brazil, Denmark, France, Mexico, in Aspergillus niger Australia and New Zealand. -

In Vitro Inhibition of HIV-1 Proteinase by Cerulenin
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 261, number 2, 373-377 FEBS 08165 February 1990 In vitro inhibition of HIV-1 proteinase by cerulenin Karin Moelling, Thomas Schulze, Marie-Theres Knoop, John Kay +, Raymond Jupp +, George Nicolaou* and Laurence H. Pearl* Max-Planck lnstitut fiir Molekular Genetik, lhnestrasse 73, D-IO00 Berlin 33, FRG, +Department of Biochemistry, University College of Wales, PO Box 903, Cardiff CFl 1ST, UK and *Department of Biochemistry, University College London, Gower Street, London WCIE 6BT, UK Received 13 October 1989; revised version received 18 December 1989 Retroviruses encode proteinases necessary for the proteolytic processing of the viral gag and gag-pol precursor proteins. These enzymes have been shown to be structurally and functionally related to aspartyl proteinases such as pepsin and renin. Cerulenin is a naturally occurring antibiotic, commonly used as an inhibitor of fatty acid synthesis. Cerulenin has been observed to inhibit production of Rous sarcoma virus and murine leukae- mia virus by infected cells, possibly by interfering with proteolytic processing of viral precursor proteins. We show here that cerulenin inhibits the action of the HIV-1 proteinase in vitro, using 3 substrates: a synthetic heptapeptide (SQNYPIV) which corresponds to the sequence at the HIV-I gag p17/p24 junction, a bacterially expressed gag precursor, and purified 66 kDa reverse transcriptase. Inhibition of cleavage by HIV-1 proteinase required preincubation with cerulenin. Cerulenin also inactivates endothiapepsin, a well-characterised fungal aspartyl proteinase, sug- gesting that the action of cerulenin is a function of the common active site structure of the retroviral and aspartic proteinases. -

Molecular Markers of Serine Protease Evolution
The EMBO Journal Vol. 20 No. 12 pp. 3036±3045, 2001 Molecular markers of serine protease evolution Maxwell M.Krem and Enrico Di Cera1 ment and specialization of the catalytic architecture should correspond to signi®cant evolutionary transitions in the Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, Box 8231, St Louis, history of protease clans. Evolutionary markers encoun- MO 63110-1093, USA tered in the sequences contributing to the catalytic apparatus would thus give an account of the history of 1Corresponding author e-mail: [email protected] an enzyme family or clan and provide for comparative analysis with other families and clans. Therefore, the use The evolutionary history of serine proteases can be of sequence markers associated with active site structure accounted for by highly conserved amino acids that generates a model for protease evolution with broad form crucial structural and chemical elements of applicability and potential for extension to other classes of the catalytic apparatus. These residues display non- enzymes. random dichotomies in either amino acid choice or The ®rst report of a sequence marker associated with serine codon usage and serve as discrete markers for active site chemistry was the observation that both AGY tracking changes in the active site environment and and TCN codons were used to encode active site serines in supporting structures. These markers categorize a variety of enzyme families (Brenner, 1988). Since serine proteases of the chymotrypsin-like, subtilisin- AGY®TCN interconversion is an uncommon event, it like and a/b-hydrolase fold clans according to phylo- was reasoned that enzymes within the same family genetic lineages, and indicate the relative ages and utilizing different active site codons belonged to different order of appearance of those lineages. -

Processing of Antigenic Peptides by Aminopeptidases
June 2004 Biol. Pharm. Bull. 27(6) 777—780 (2004) 777 Current Topics Aminopeptidases in Health and Disease Processing of Antigenic Peptides by Aminopeptidases Akira HATTORI* and Masafumi TSUJIMOTO Laboratory of Cellular Biochemistry, RIKEN; 2–1 Hirosawa, Wako, Saitama 351–0198, Japan. Received January 7, 2004 Antigenic peptides presented to major histocompatibility complex (MHC) class I molecules are generated in the cytosol during degradation of cellular proteins by the ubiquitin-proteasome proteolytic pathway. Proteasome can generate N-extended precursors as well as final epitopes, and then the precursors are processed to mature epitopes by aminopeptidases. Both cytosolic peptidases (i.e. puromycin-sensitive aminopeptidase, bleomycin hy- drolase and interferon-g-inducible leucine aminopeptidase) and recently identified metallo-aminopeptidase lo- cated in the endoplasmic reticulum (i.e. adipocyte-derived leucine aminopeptidase/endoplasmic reticulum aminopeptidase 1 and leukocyte-derived arginine aminopeptidase) can generate final epitopes from precursor peptides. Some of these aminopeptidases are also considered to destroy certain antigenic peptides to limit the antigen presentation. Taken together, it is getting evident that aminopeptidases located in the cytosol and the lumen of endoplasmic reticulum play important roles in the generation of antigenic peptides presented to MHC class I molecules. Key words aminopeptidase; antigen processing; major histocompatibility complex (MHC) class I; antigen presentation; protea- some; protein degradation -

Membrane Proteins • Cofactors – Plimstex • Membranes • Dna • Small Molecules/Gas • Large Complexes
Structural mass spectrometry hydrogen/deuterium exchange Petr Man Structural Biology and Cell Signalling Institute of Microbiology, Czech Academy of Sciences Structural biology methods Low-resolution methods High-resolution methods Rigid SAXS IR Raman CD ITC MST Cryo-EM AUC SPR MS X-ray crystallography Chemical cross-linking H/D exchange Native ESI + ion mobility Oxidative labelling Small Large NMR Dynamic Structural biology approaches Simple MS, quantitative MS Cross-linking, top-down, native MS+dissociation native MS+ion mobility Cross-linking Structural MS What can we get using mass spectrometry IM – ion mobility CXL – chemical cross-linking AP – afinity purification OFP – oxidative footprinting HDX – hydrogen/deuterium exchange ISOTOPE EXCHANGE IN PROTEINS 1H 2H 3H occurence [%] 99.988 0.0115 trace 5 …Kaj Ulrik Linderstrøm-Lang „Cartesian diver“ Proteins are migrating in tubes with density gradient until they stop at the point where the densities are equal 1H 2H 3H % 99.9885 0.0115 trace density [g/cm3] 1.000 1.106 1.215 Methods of detection IR: β-: NMR: 1 n = 1.6749 × 10-27 kg MS: 1H 2H 3H výskyt% [%] 99.9885 0.0115 trace hustotadensity vody [g/cm [g/cm3] 3] 1.000 1.106 1.215 jadernýspinspin ½+ 1+ ½+ mass [u] 1.00783 2.01410 3.01605 Factors affecting H/D exchange hydrogen bonding solvent accessibility Factors affecting H/D exchange Side chains (acidity, steric shielding) Bai et al.: Proteins (1993) Glasoe, Long: J. Phys. Chem. (1960) Factors affecting H/D exchange – side chain effects Inductive effect – electron density is Downward shift due to withdrawn from peptide steric hindrance effect of bond (S, O). -

(12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS Et Al
US 2016.0346364A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS et al. (43) Pub. Date: Dec. 1, 2016 (54) MEDICAMENT AND METHOD FOR (52) U.S. Cl. TREATING INNATE IMMUNE RESPONSE CPC ........... A61K 38/488 (2013.01); A61K 38/482 DISEASES (2013.01); C12Y 304/23019 (2013.01); C12Y 304/21026 (2013.01); C12Y 304/23018 (71) Applicant: DSM IPASSETS B.V., Heerlen (NL) (2013.01); A61K 9/0053 (2013.01); C12N 9/62 (2013.01); A23L 29/06 (2016.08); A2ID 8/042 (72) Inventors: Maaike Johanna BRUINS, Kaiseraugst (2013.01); A23L 5/25 (2016.08); A23V (CH); Luppo EDENS, Kaiseraugst 2002/00 (2013.01) (CH); Lenneke NAN, Kaiseraugst (CH) (57) ABSTRACT (21) Appl. No.: 15/101,630 This invention relates to a medicament or a dietary Supple (22) PCT Filed: Dec. 11, 2014 ment comprising the Aspergillus niger aspergilloglutamic peptidase that is capable of hydrolyzing plant food allergens, (86). PCT No.: PCT/EP2014/077355 and more particularly, alpha-amylase/trypsin inhibitors, thereby treating diseases due to an innate immune response S 371 (c)(1), in humans, and/or allowing to delay the onset of said (2) Date: Jun. 3, 2016 diseases. The present invention relates to the discovery that (30) Foreign Application Priority Data the Aspergillus niger aspergilloglutamic peptidase is capable of hydrolyzing alpha-amylase/trypsin inhibitors that are Dec. 11, 2013 (EP) .................................. 13196580.8 present in wheat and related cereals said inhibitors being strong inducers of innate immune response. Furthermore, Publication Classification the present invention relates to a method for hydrolyzing alpha-amylase/trypsin inhibitors comprising incubating a (51) Int. -

Structure-Based Optimization of Inhibitors of the Aspartic Protease Endothiapepsin
Int. J. Mol. Sci. 2015, 16, 19184-19194; doi:10.3390/ijms160819184 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Structure-Based Optimization of Inhibitors of the Aspartic Protease Endothiapepsin Alwin M. Hartman 1,†, Milon Mondal 1,†, Nedyalka Radeva 2, Gerhard Klebe 2 and Anna K. H. Hirsch 1,* 1 Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands; E-Mails: [email protected] (A.M.H.); [email protected] (M.M.) 2 Institute of Pharmaceutical Chemistry, Philipps-University Marburg, Marbacher Weg 6, 35032 Marburg, Germany; E-Mails: [email protected] (N.R.); [email protected] (G.K.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Fax: +31-50-363-4296. Academic Editor: John George Hardy Received: 1 May 2015 / Accepted: 6 July 2015 / Published: 14 August 2015 Abstract: Aspartic proteases are a class of enzymes that play a causative role in numerous diseases such as malaria (plasmepsins), Alzheimer’s disease (β-secretase), fungal infections (secreted aspartic proteases), and hypertension (renin). We have chosen endothiapepsin as a model enzyme of this class of enzymes, for the design, preparation and biochemical evaluation of a new series of inhibitors of endothiapepsin. Here, we have optimized a hit, identified by de novo structure-based drug design (SBDD) and DCC, by using structure-based design approaches focusing on the optimization of an amide–π interaction. Biochemical results are in agreement with SBDD. -

Ubiquitin-Mediated Proteolysis the Nobel Prize in Chemistry for 2004 Is
Advanced information on the Nobel Prize in Chemistry, 6 October 2004 Information Department, P.O. Box 50005, SE-104 05 Stockholm, Sweden Phone: +46 8 673 95 00, Fax: +46 8 15 56 70, E-mail: [email protected], Website: www.kva.se Ubiquitin-mediated proteolysis The Nobel Prize in Chemistry for 2004 is shared between three scientists who have made fundamental discoveries concerning how cells regulate the breakdown of intracellular proteins with extreme specificity as to target, time and space. Aaron Ciechanover, Avram Hershko and Irwin Rose together discovered ubiquitin- mediated proteolysis, a process where an enzyme system tags unwanted proteins with many molecules of the 76-amino acid residue protein ubiquitin. The tagged proteins are then transported to the proteasome, a large multisubunit protease complex, where they are degraded. Numerous cellular processes regulated by ubiquitin-mediated proteolysis include the cell cycle, DNA repair and transcription, protein quality control and the immune response. Defects in this proteolysis have a causal role in many human diseases, including a variety of cancers. Fig. 1 Ubiquitin-mediated proteolysis and its many biological functions 2 Introduction Eukaryotic cells, from yeast to human, contain some 6000 to 30000 protein-encoding genes and at least as many proteins. While much attention and research had been devoted to how proteins are synthesized, the reverse process, i.e. how proteins are degraded, long received little attention. A pioneer in this field was Schoenheimer, who in 1942 published results from isotope tracer techniques indicating that proteins in animals are continuously synthesized and degraded and therefore are in a dynamic state (Schoenheimer, 1942). -

Progress in the Field of Aspartic Proteinases in Cheese Manufacturing
Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering Sirma Yegin, Peter Dekker To cite this version: Sirma Yegin, Peter Dekker. Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering. Dairy Science & Technology, EDP sciences/Springer, 2013, 93 (6), pp.565-594. 10.1007/s13594-013-0137-2. hal-01201447 HAL Id: hal-01201447 https://hal.archives-ouvertes.fr/hal-01201447 Submitted on 17 Sep 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Dairy Sci. & Technol. (2013) 93:565–594 DOI 10.1007/s13594-013-0137-2 REVIEW PAPER Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering Sirma Yegin & Peter Dekker Received: 25 February 2013 /Revised: 16 May 2013 /Accepted: 21 May 2013 / Published online: 27 June 2013 # INRA and Springer-Verlag France 2013 Abstract Aspartic proteinases are an important class of proteinases which are widely used as milk-coagulating agents in industrial cheese production. They are available from a wide range of sources including mammals, plants, and microorganisms. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Crystal Structures of Native and Inhibitedforms of Human Cathepsin
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 6796-6800, July 1993 Biochemustry Crystal structures of native and inhibited forms of human cathepsin D: Implications for lysosomal targeting and drug design (aspartic protcase/N-linked oligosaccharide/pepstatin A) ERic T. BALDWIN*, T. NARAYANA BHAT*, SERGEI GULNIK*, MADHUSOODAN V. HOSUR*t, RAYMOND C. SOWDER Il, RAUL E. CACHAU*, JACK COLLINS*, ABELARDO M. SILVA*, AND JOHN W. ERICKSON*§ *Structural Biochemistry Program, Frederick Biomedical Supercomputing Center and tAIDS Vaccine Program, Program Resources Inc./DynCorp, National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD 21702 Communicated by David R. Davies, March 24, 1993 (receivedfor review February 4, 1993) ABSTRACT Cathepsin D (EC 3.4.23.5) is a lysosomal duced in the vicinity of the growing tumor, may degrade the protease suspected to play important roles in protein catabo- extracellular matrix and thereby promote the escape of lism, antigen processing, degenerative diseases, and breast cancer cells to the lymphatic and circulatory systems and cancer progresson. Determination of the crystal structures of enhance the invasion of new tissues (17, 18). The design of cathepsin D and a complex with pepstatin at 2.5 A resolution potent and specific inhibitors of cathepsin D will aid the provides insights into inhibitor binding and lysosomal targeting further elucidation of the roles of this enzyme in human for this two-chain, N-glycosylated aspartic protease. Compar- disease. We previously described the purification and crys- ison with the structures of a complex of pepstatin bound to tallization ofhuman cathepsin D from liver (3); similar studies rhizopuspepsin and with a human renin-bihbitor complex have been reported recently for cathepsin D isolated from revealed differences in subsite structures and inhibitor-enzyme bovine liver (19) and human spleen (20). -

BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’S Disease Pathology
Review BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’s Disease Pathology Gerald Koelsch CoMentis, Inc., South San Francisco, CA 94080, USA; [email protected] Received: 15 September 2017; Accepted: 10 October 2017; Published: 13 October 2017 Abstract: Alzheimer’s disease (AD) is a fatal progressive neurodegenerative disorder characterized by increasing loss in memory, cognition, and function of daily living. Among the many pathologic events observed in the progression of AD, changes in amyloid β peptide (Aβ) metabolism proceed fastest, and precede clinical symptoms. BACE1 (β-secretase 1) catalyzes the initial cleavage of the amyloid precursor protein to generate Aβ. Therefore inhibition of BACE1 activity could block one of the earliest pathologic events in AD. However, therapeutic BACE1 inhibition to block Aβ production may need to be balanced with possible effects that might result from diminished physiologic functions BACE1, in particular processing of substrates involved in neuronal function of the brain and periphery. Potentials for beneficial or consequential effects resulting from pharmacologic inhibition of BACE1 are reviewed in context of ongoing clinical trials testing the effect of BACE1 candidate inhibitor drugs in AD populations. Keywords: Alzheimer’s disease; amyloid hypothesis; BACE1; beta secretase; pharmacology 1. Introduction Alzheimer’s disease (AD) is a fatal progressive neurodegenerative disorder, slowly eroding memory, cognition, and functions of daily living, inevitably culminating in death from pneumonia and infectious diseases resulting from failure to thrive, loss of fine motor skills, and incapacitation. Treatment is limited to therapeutics that alleviate symptoms of memory loss, but are effective for a relatively short duration during and after which disease progression continues.