JASON MARC GOLDSTEIN the Isolation, Characterization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Markers of Serine Protease Evolution
The EMBO Journal Vol. 20 No. 12 pp. 3036±3045, 2001 Molecular markers of serine protease evolution Maxwell M.Krem and Enrico Di Cera1 ment and specialization of the catalytic architecture should correspond to signi®cant evolutionary transitions in the Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, Box 8231, St Louis, history of protease clans. Evolutionary markers encoun- MO 63110-1093, USA tered in the sequences contributing to the catalytic apparatus would thus give an account of the history of 1Corresponding author e-mail: [email protected] an enzyme family or clan and provide for comparative analysis with other families and clans. Therefore, the use The evolutionary history of serine proteases can be of sequence markers associated with active site structure accounted for by highly conserved amino acids that generates a model for protease evolution with broad form crucial structural and chemical elements of applicability and potential for extension to other classes of the catalytic apparatus. These residues display non- enzymes. random dichotomies in either amino acid choice or The ®rst report of a sequence marker associated with serine codon usage and serve as discrete markers for active site chemistry was the observation that both AGY tracking changes in the active site environment and and TCN codons were used to encode active site serines in supporting structures. These markers categorize a variety of enzyme families (Brenner, 1988). Since serine proteases of the chymotrypsin-like, subtilisin- AGY®TCN interconversion is an uncommon event, it like and a/b-hydrolase fold clans according to phylo- was reasoned that enzymes within the same family genetic lineages, and indicate the relative ages and utilizing different active site codons belonged to different order of appearance of those lineages. -

Processing of Antigenic Peptides by Aminopeptidases
June 2004 Biol. Pharm. Bull. 27(6) 777—780 (2004) 777 Current Topics Aminopeptidases in Health and Disease Processing of Antigenic Peptides by Aminopeptidases Akira HATTORI* and Masafumi TSUJIMOTO Laboratory of Cellular Biochemistry, RIKEN; 2–1 Hirosawa, Wako, Saitama 351–0198, Japan. Received January 7, 2004 Antigenic peptides presented to major histocompatibility complex (MHC) class I molecules are generated in the cytosol during degradation of cellular proteins by the ubiquitin-proteasome proteolytic pathway. Proteasome can generate N-extended precursors as well as final epitopes, and then the precursors are processed to mature epitopes by aminopeptidases. Both cytosolic peptidases (i.e. puromycin-sensitive aminopeptidase, bleomycin hy- drolase and interferon-g-inducible leucine aminopeptidase) and recently identified metallo-aminopeptidase lo- cated in the endoplasmic reticulum (i.e. adipocyte-derived leucine aminopeptidase/endoplasmic reticulum aminopeptidase 1 and leukocyte-derived arginine aminopeptidase) can generate final epitopes from precursor peptides. Some of these aminopeptidases are also considered to destroy certain antigenic peptides to limit the antigen presentation. Taken together, it is getting evident that aminopeptidases located in the cytosol and the lumen of endoplasmic reticulum play important roles in the generation of antigenic peptides presented to MHC class I molecules. Key words aminopeptidase; antigen processing; major histocompatibility complex (MHC) class I; antigen presentation; protea- some; protein degradation -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Enzymatic Hydrolysis and Peptide Mapping of Potato Pulp Protein
Enzymatic Hydrolysis and Peptide Mapping of Potato Pulp Protein Von der Naturwissenschaftlichen Fakultät der Universität Hannover zur Erlangung des Grades Doktorin der Naturwissenschaften Dr. rer. nat. genehmigte Dissertation von Chulaporn Kamnerdpetch, M.Sc. geboren in Bangkok, Thailand Hannover 2006 Hauptreferent Prof. Dr. Thomas Scheper Institut für Technische Chemie Universität Hannover Koreferent Prof. Dr. Bernd Hitzmann Institut für Technische Chemie Universität Hannover Tag der Promotion 29. Mai 2006 Erklärung Ich versichere, dass ich diese Dissertation selbstständig und nur unter Verwendung der angegebenen Hilfsmittel und Quellen durchgeführt habe. Diese Arbeit wurde nicht als Diplomarbeit oder ähnliche Prüfungsarbeit verwendet. Chulaporn Kamnerdpetch Hannover, den 29. Mai 2006 ACKNOWLEDGEMENTS This thesis is the result of my four years research work whereby I have been accompanied and supported by many people. It is a pleasant aspect that I have now the opportunity to express my sincere gratitude for all of them who made this thesis possible. The first person I would like to thank is my supervisor Prof. Dr. Thomas Scheper for giving me the opportunity to take part on the doctoral program at the Institut für Technische Chemie der Universität Hannover. I appreciate very much for his enthusiastic and enthusing support. He gave me an encourage independent thinking and the freedom to try out my ways. I would like to thank to Prof. Dr. Bernd Hitzmann for his kindness acceptance as my co-referee. I wish to express my thank to Dr. Cornelia Kasper for preparing my publication and proof reading. It is a great pleasure for me to thank Dr. Pichai Namparkai for proof reading as well. -

High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma
molecules Article High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma Elisa Maffioli 1,2 , Zhenze Jiang 3, Simona Nonnis 1,2 , Armando Negri 1,2, Valentina Romeo 1, Christopher B. Lietz 3, Vivian Hook 3,4, Giuseppe Ristagno 5, Giuseppe Baselli 6, Erik B. Kistler 7,8 , Federico Aletti 9, Anthony J. O’Donoghue 3,* and Gabriella Tedeschi 1,2,* 1 Department of Veterinary Medicine, University of Milano, 20133 Milano, Italy; elisa.maffi[email protected] (E.M.); [email protected] (S.N.); [email protected] (A.N.); [email protected] (V.R.) 2 Centre for Nanostructured Materials and Interfaces (CIMAINA), University of Milano, 20133 Milano, Italy 3 Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA 92093, USA; [email protected] (Z.J.); [email protected] (C.B.L.); [email protected] (V.H.) 4 Department of Neurosciences, School of Medicine, University of California San Diego, La Jolla, CA 92093, USA 5 Department of Pathophysiology and Transplantation, University of Milan, 20133 Milan, Italy; [email protected] 6 Dipartimento di Elettronica, Informazione e Bioingegneria, Politecnico di Milano, 20133 Milan, Italy; [email protected] 7 Department of Anesthesiology & Critical Care, University of California San Diego, La Jolla, CA 92093, USA; [email protected] 8 Department of Anesthesiology & Critical Care, VA San Diego HealthCare System, San Diego, CA 92161, USA 9 Department of Bioengineering, University of California San Diego, La Jolla, CA 92093, USA; [email protected] * Correspondence: [email protected] (A.J.O.); [email protected] (G.T.); Tel.: +1-8585345360 (A.J.O.); +39-02-50318127 (G.T.) Academic Editor: Paolo Iadarola Received: 28 July 2020; Accepted: 4 September 2020; Published: 6 September 2020 Abstract: Proteomic technologies have identified 234 peptidases in plasma but little quantitative information about the proteolytic activity has been uncovered. -

(12) Patent Application Publication (10) Pub. No.: US 2006/0110747 A1 Ramseier Et Al
US 200601 10747A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0110747 A1 Ramseier et al. (43) Pub. Date: May 25, 2006 (54) PROCESS FOR IMPROVED PROTEIN (60) Provisional application No. 60/591489, filed on Jul. EXPRESSION BY STRAIN ENGINEERING 26, 2004. (75) Inventors: Thomas M. Ramseier, Poway, CA Publication Classification (US); Hongfan Jin, San Diego, CA (51) Int. Cl. (US); Charles H. Squires, Poway, CA CI2O I/68 (2006.01) (US) GOIN 33/53 (2006.01) CI2N 15/74 (2006.01) Correspondence Address: (52) U.S. Cl. ................................ 435/6: 435/7.1; 435/471 KING & SPALDING LLP 118O PEACHTREE STREET (57) ABSTRACT ATLANTA, GA 30309 (US) This invention is a process for improving the production levels of recombinant proteins or peptides or improving the (73) Assignee: Dow Global Technologies Inc., Midland, level of active recombinant proteins or peptides expressed in MI (US) host cells. The invention is a process of comparing two genetic profiles of a cell that expresses a recombinant (21) Appl. No.: 11/189,375 protein and modifying the cell to change the expression of a gene product that is upregulated in response to the recom (22) Filed: Jul. 26, 2005 binant protein expression. The process can improve protein production or can improve protein quality, for example, by Related U.S. Application Data increasing solubility of a recombinant protein. Patent Application Publication May 25, 2006 Sheet 1 of 15 US 2006/0110747 A1 Figure 1 09 010909070£020\,0 10°0 Patent Application Publication May 25, 2006 Sheet 2 of 15 US 2006/0110747 A1 Figure 2 Ester sers Custer || || || || || HH-I-H 1 H4 s a cisiers TT closers | | | | | | Ya S T RXFO 1961. -

Proteasome Inhibitors: from Research Tools to Drug Candidates
Chemistry & Biology 8 (2001) 739^758 www.elsevier.com/locate/chembiol Review Proteasome inhibitors: from research tools to drug candidates Alexei F. Kisselev*, Alfred L. Goldberg Department of Cell Biology, Harvard Medical School, 240 Longwood Ave., Boston, MA 02115, USA Received 13 December 2000; revisions requested 29 March 2001; revisions received 12 June 2001; accepted 19 June 2001 First published online 12 July 2001 Abstract The 26S proteasome is a 2.4 MDa multifunctional ATP- inhibitors are now in clinical trials for treatment of multiple dependent proteolytic complex, which degrades the majority of cancers and stroke. ß 2001 Elsevier Science Ltd. All rights re- cellular polypeptides by an unusual enzyme mechanism. Several served. groups of proteasome inhibitors have been developed and are now widely used as research tools to study the role of the ubiquitin^ Keywords: Proteasome inhibitor; proteasome pathway in various cellular processes, and two Ubiquitin^proteasome pathway; Drug candidate 1. Introduction of cyclins and inhibitors of cyclin-dependent kinases [7], while degradation of transcriptional regulators, such as The ubiquitin^proteasome pathway is the major proteo- c-Jun, E2F-1 and L-catenin (see [8] for review) is essential lytic system in the cytosol and nucleus of all eukaryotic for the regulation of cell growth and gene expression. cells. This ATP-dependent pathway was discovered more Similarly, degradation by the proteasome of activated pro- than 20 years ago [1,2], but the involvement of the pro- tein kinases, e.g. src and protein kinase C [9,10], is critical teasome particle was demonstrated only in the late 1980s for the termination of certain signal transduction cascades. -

Studies of Structure and Function of Tripeptidyl-Peptidase II
Till familj och vänner List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I. Eriksson, S.; Gutiérrez, O.A.; Bjerling, P.; Tomkinson, B. (2009) De- velopment, evaluation and application of tripeptidyl-peptidase II se- quence signatures. Archives of Biochemistry and Biophysics, 484(1):39-45 II. Lindås, A-C.; Eriksson, S.; Josza, E.; Tomkinson, B. (2008) Investiga- tion of a role for Glu-331 and Glu-305 in substrate binding of tripepti- dyl-peptidase II. Biochimica et Biophysica Acta, 1784(12):1899-1907 III. Eklund, S.; Lindås, A-C.; Hamnevik, E.; Widersten, M.; Tomkinson, B. Inter-species variation in the pH dependence of tripeptidyl- peptidase II. Manuscript IV. Eklund, S.; Kalbacher, H.; Tomkinson, B. Characterization of the endopeptidase activity of tripeptidyl-peptidase II. Manuscript Paper I and II were published under maiden name (Eriksson). Reprints were made with permission from the respective publishers. Contents Introduction ..................................................................................................... 9 Enzymes ..................................................................................................... 9 Enzymes and pH dependence .............................................................. 11 Peptidases ................................................................................................. 12 Serine peptidases ................................................................................. 14 Intracellular protein -

The Role of Tricorn Protease and Its Aminopeptidase-Interacting Factors in Cellular Protein Degradation
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Cell, Vol. 95, 637±648, November 25, 1998, Copyright 1998 by Cell Press The Role of Tricorn Protease and Its Aminopeptidase-Interacting Factors in Cellular Protein Degradation Noriko Tamura, Friedrich Lottspeich, complexes that invariably contain ATPase subunits ren- Wolfgang Baumeister,* and Tomohiro Tamura dering protein degradation energy dependent. The oc- Max-Planck-Institut fuÈ r Biochemie currence of self-compartmentalizing proteases in all D-82152 Martinsried three domains of life bears testimony of an old evolution- Germany ary principle. In prokaryotic cells lacking membrane- bounded compartments, ATP-dependent self-compart- mentalizing proteases such as the proteasome, HslV, ClpP, or Lon are responsible for the bulk of the protein Summary turnover. Beyond facilitating the control of proteolysis, the con- Tricorn protease was previously described as the core finement of the proteolytic activity to a nanocompart- enzyme of a modular proteolytic system displaying ment inside these assemblies also provides the struc- multicatalytic activity. Here we elucidate the mode of tural basis for the processive mode of action that is cooperation between Tricorn and its interacting fac- characteristic for these proteases: they do not release tors, and we identify two additional factors, F2 and F3, fragments after a single cleavage, but proceed to make closely related aminopeptidases of 89 kDa. In conjunc- multiple cleavages before finally discharging the degra- tion with these three factors, Tricorn degrades oligo- dation products (Thompson et al., 1994; Akopian et al., peptides in a sequential manner, yielding free amino 1997). -
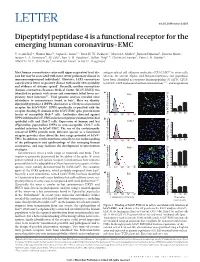
Dipeptidyl Peptidase 4 Is a Functional Receptor for the Emerging Human Coronavirus-EMC
LETTER doi:10.1038/nature12005 Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC V. Stalin Raj1*, Huihui Mou2*, Saskia L. Smits1,3, Dick H. W. Dekkers4, Marcel A. Mu¨ller5, Ronald Dijkman6, Doreen Muth5, Jeroen A. A. Demmers4, Ali Zaki7, Ron A. M. Fouchier1, Volker Thiel6,8, Christian Drosten5, Peter J. M. Rottier2, Albert D. M. E. Osterhaus1, Berend Jan Bosch2 & Bart L. Haagmans1 Most human coronaviruses cause mild upper respiratory tract dis- antigen-related cell adhesion molecules (CEACAM)10 to enter cells, ease but may be associated with more severe pulmonary disease in whereas for several Alpha- and Betacoronaviruses, two peptidases immunocompromised individuals1. However, SARS coronavirus have been identified as receptors (aminopeptidase N (APN, CD13) caused severe lower respiratory disease with nearly 10% mortality for hCoV-229E and several animal coronaviruses11,12, and angiotensin and evidence of systemic spread2. Recently, another coronavirus (human coronavirus-Erasmus Medical Center (hCoV-EMC)) was a identified in patients with severe and sometimes lethal lower res- )] Vero –1 piratory tract infection3,4. Viral genome analysis revealed close 9 ml 8 5 50 relatedness to coronaviruses found in bats . Here we identify 7 dipeptidyl peptidase 4 (DPP4; also known as CD26) as a functional 6 5 receptor for hCoV-EMC. DPP4 specifically co-purified with the 4 receptor-binding S1 domain of the hCoV-EMC spike protein from 3 Relative cell number log[GE (TCID 0 20 40 lysates of susceptible Huh-7 cells. Antibodies directed against 0 101 102 103 104 DPP4 inhibited hCoV-EMC infection of primary human bronchial b )] COS-7 –1 epithelial cells and Huh-7 cells. -

And Exopeptidases in a Processing Enzyme System: Activation
Proc. Nail. Acad. Sci. USA Vol. 85, pp. 5468-5472, August 1988 Biochemistry Relationship between endo- and exopeptidases in a processing enzyme system: Activation of an endoprotease by the aminopeptidase B-like activity in somatostatin-28 convertase (brain cortex/basic amino acid pairs/peptide substrates/protease inhibitors/prohormone maturation) SOPHIE GOMEZ, PABLO GLUSCHANKOF, AGNES LEPAGE, AND PAUL COHEN Groupe de Neurobiochimie Cellulaire et Moldculaire, Universitd Pierre et Marie Curie, Unitd Associde 554 au Centre National de la Recherche Scientifique, 96 boulevard Raspail, 75006 Paris, France Communicated by I. Robert Lehman, April 8, 1988 (receivedfor review December 15, 1987) ABSTRACT The somatostatin-28 convertase activity in- somatostatin-14 and the amino-terminal dodecapeptide so- volved in vitro in the processing of somatostatin-28 into the matostatin-28-(1-12) (16). neuropeptides somatostatin-28-(1-12) and somatostatin-14 is We have described an endoprotease that cleaves the composed of an endoprotease and a basic aminopeptidase. We peptide bond on the amino side ofthe Arg-Lys doublet in the report herein on the purification to apparent homogeneity of somatostatin-28 sequence (17, 18), releasing the somato- these two constituents and on their functional interrelationship. statin-28-(1-12) fragment and [Arg-2,Lys-1]somatostatin-14. In particular we observed that after various physicochemical The released [Arg-2,Lys-']somatostatin-14 is further proc- treatments, the 90-kDa endoprotease activity was recovered essed by an aminopeptidase B-like activity (18, 19) that is also both at this molecular mass and as a 45-kDa entity. Moreover, present in the preparation. We report herein the purification the production of [Arg2,LysJllsomatostatin-14 from somato- to apparent homogeneity ofthese two activities. -

Proteolytic Cleavage—Mechanisms, Function
Review Cite This: Chem. Rev. 2018, 118, 1137−1168 pubs.acs.org/CR Proteolytic CleavageMechanisms, Function, and “Omic” Approaches for a Near-Ubiquitous Posttranslational Modification Theo Klein,†,⊥ Ulrich Eckhard,†,§ Antoine Dufour,†,¶ Nestor Solis,† and Christopher M. Overall*,†,‡ † ‡ Life Sciences Institute, Department of Oral Biological and Medical Sciences, and Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada ABSTRACT: Proteases enzymatically hydrolyze peptide bonds in substrate proteins, resulting in a widespread, irreversible posttranslational modification of the protein’s structure and biological function. Often regarded as a mere degradative mechanism in destruction of proteins or turnover in maintaining physiological homeostasis, recent research in the field of degradomics has led to the recognition of two main yet unexpected concepts. First, that targeted, limited proteolytic cleavage events by a wide repertoire of proteases are pivotal regulators of most, if not all, physiological and pathological processes. Second, an unexpected in vivo abundance of stable cleaved proteins revealed pervasive, functionally relevant protein processing in normal and diseased tissuefrom 40 to 70% of proteins also occur in vivo as distinct stable proteoforms with undocumented N- or C- termini, meaning these proteoforms are stable functional cleavage products, most with unknown functional implications. In this Review, we discuss the structural biology aspects and mechanisms