Adaptively Weighted and Robust Mathematical Programming for the Discovery of Driver Gene Sets in Cancers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Identification of DNA Response Elements Regulating Expression of CCAAT/Enhancer-Binding Protein (C/EBP) Β and Δ During Early A
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.22.110114; this version posted May 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Identification of DNA response elements regulating expression of CCAAT/enhancer-binding protein (C/EBP) β and ẟ during early adipogenesis James E. Merrett1,2, Tao Bo1,4, Peter J. Psaltis1,3 and Christopher G. Proud1,2 * 1South Australian Health and Medical Research Institute, Lifelong Health, Adelaide, South Australia, Australia 2University of Adelaide, Department of Molecular and Biomedical Science, Adelaide, South Australia, Australia 3University of Adelaide, Adelaide Medical School, Adelaide, South Australia, Australia 4Shandong-South Australia Joint Laboratory of Metabolic Disease Research, Shandong Provincial Hospital affiliated to Shandong First Medical University *Corresponding author: Christopher Proud E-mail: [email protected] Running title: DNA response elements regulating early adipogenesis Keywords: adipogenesis, adipocyte, cyclic AMP (cAMP), cAMP response element-binding protein (CREB), CCAAT/enhancer-binding protein (C/EBP), glucocorticoid, glucocorticoid receptor, MAPK- interacting kinase (MNK) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.22.110114; this version posted May 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Given the high and increasing prevalence of obesity and associated disorders, such as type-2 diabetes, it is important to understand the mechanisms that regulate lipid storage and the differentiation of fat cells, a process termed adipogenesis. -

Supplementary Table 1
SI Table S1. Broad protein kinase selectivity for PF-2771. Kinase, PF-2771 % Inhibition at 10 μM Service Kinase, PF-2771 % Inhibition at 1 μM Service rat RPS6KA1 (RSK1) 39 Dundee AURKA (AURA) 24 Invitrogen IKBKB (IKKb) 26 Dundee CDK2 /CyclinA 21 Invitrogen mouse LCK 25 Dundee rabbit MAP2K1 (MEK1) 19 Dundee AKT1 (AKT) 21 Dundee IKBKB (IKKb) 16 Dundee CAMK1 (CaMK1a) 19 Dundee PKN2 (PRK2) 14 Dundee RPS6KA5 (MSK1) 18 Dundee MAPKAPK5 14 Dundee PRKD1 (PKD1) 13 Dundee PIM3 12 Dundee MKNK2 (MNK2) 12 Dundee PRKD1 (PKD1) 12 Dundee MARK3 10 Dundee NTRK1 (TRKA) 12 Invitrogen SRPK1 9 Dundee MAPK12 (p38g) 11 Dundee MAPKAPK5 9 Dundee MAPK8 (JNK1a) 11 Dundee MAPK13 (p38d) 8 Dundee rat PRKAA2 (AMPKa2) 11 Dundee AURKB (AURB) 5 Dundee NEK2 11 Invitrogen CSK 5 Dundee CHEK2 (CHK2) 11 Invitrogen EEF2K (EEF-2 kinase) 4 Dundee MAPK9 (JNK2) 9 Dundee PRKCA (PKCa) 4 Dundee rat RPS6KA1 (RSK1) 8 Dundee rat PRKAA2 (AMPKa2) 4 Dundee DYRK2 7 Dundee rat CSNK1D (CKId) 3 Dundee AKT1 (AKT) 7 Dundee LYN 3 BioPrint PIM2 7 Invitrogen CSNK2A1 (CKIIa) 3 Dundee MAPK15 (ERK7) 6 Dundee CAMKK2 (CAMKKB) 1 Dundee mouse LCK 5 Dundee PIM3 1 Dundee PDPK1 (PDK1) (directed 5 Invitrogen rat DYRK1A (MNB) 1 Dundee RPS6KB1 (p70S6K) 5 Dundee PBK 0 Dundee CSNK2A1 (CKIIa) 4 Dundee PIM1 -1 Dundee CAMKK2 (CAMKKB) 4 Dundee DYRK2 -2 Dundee SRC 4 Invitrogen MAPK12 (p38g) -2 Dundee MYLK2 (MLCK_sk) 3 Invitrogen NEK6 -3 Dundee MKNK2 (MNK2) 2 Dundee RPS6KB1 (p70S6K) -3 Dundee SRPK1 2 Dundee AKT2 -3 Dundee MKNK1 (MNK1) 2 Dundee RPS6KA3 (RSK2) -3 Dundee CHEK1 (CHK1) 2 Invitrogen rabbit MAP2K1 (MEK1) -4 Dundee -

Gene Signature of the Post-Chernobyl Papillary Thyroid Cancer
CORE Metadata, citation and similar papers at core.ac.uk Provided by Spiral - Imperial College Digital Repository Eur J Nucl Med Mol Imaging DOI 10.1007/s00259-015-3303-3 ORIGINAL ARTICLE Gene signature of the post-Chernobyl papillary thyroid cancer Daria Handkiewicz-Junak1 & Michal Swierniak1,2 & Dagmara Rusinek1 & Małgorzata Oczko-Wojciechowska1 & Genevieve Dom 3 & Carine Maenhaut3 & Kristian Unger4,5 & Vincent Detours 3 & Tetiana Bogdanova 6 & Geraldine Thomas4 & Ilya Likhtarov7 & Roman Jaksik8 & Malgorzata Kowalska1 & Ewa Chmielik9 & Michal Jarzab10 & Andrzej Swierniak11 & Barbara Jarzab1 Received: 11 August 2015 /Accepted: 29 December 2015 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Human Genome U133 2.0 Plus) with the aim of identifying Purpose Following the nuclear accidents in Chernobyl and molecular differences between radiation-induced (exposed to later in Fukushima, the nuclear community has been faced Chernobyl radiation, ECR) and sporadic PTC. All participants with important issues concerning how to search for and diag- were resident in the same region so that confounding factors nose biological consequences of low-dose internal radiation related to genetics or environment were minimized. contamination. Although after the Chernobyl accident an in- Results There were small but significant differences in the crease in childhood papillary thyroid cancer (PTC) was ob- gene expression profiles between ECR and non-ECR PTC served, it is still not clear whether the molecular biology of (global test, p < 0.01), with 300 differently expressed probe PTCs associated with low-dose radiation exposure differs sets (p < 0.001) corresponding to 239 genes. Multifactorial from that of sporadic PTC. -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain -

Evolution and Structure of Clinically Relevant Gene Fusions in Multiple Myeloma
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2020 Evolution and structure of clinically relevant gene fusions in multiple myeloma Steven M. Foltz Qingsong Gao Christopher J. Yoon Hua Sun Lijun Yao See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Authors Steven M. Foltz, Qingsong Gao, Christopher J. Yoon, Hua Sun, Lijun Yao, Yize Li, Reyka G. Jayasinghe, Song Cao, Justin King, Daniel R. Kohnen, Mark A. Fiala, Li Ding, and Ravi Vij ARTICLE https://doi.org/10.1038/s41467-020-16434-y OPEN Evolution and structure of clinically relevant gene fusions in multiple myeloma Steven M. Foltz 1,2, Qingsong Gao 1,2, Christopher J. Yoon1,2, Hua Sun1,2, Lijun Yao1,2, Yize Li1,2, ✉ ✉ Reyka G. Jayasinghe1,2, Song Cao1,2, Justin King1, Daniel R. Kohnen1, Mark A. Fiala1, Li Ding1,2,3,4 & Ravi Vij1,4 Multiple myeloma is a plasma cell blood cancer with frequent chromosomal translocations leading to gene fusions. To determine the clinical relevance of fusion events, we detect gene 1234567890():,; fusions from a cohort of 742 patients from the Multiple Myeloma Research Foundation CoMMpass Study. Patients with multiple clinic visits enable us to track tumor and fusion evolution, and cases with matching peripheral blood and bone marrow samples allow us to evaluate the concordance of fusion calls in patients with high tumor burden. We examine the joint upregulation of WHSC1 and FGFR3 in samples with t(4;14)-related fusions, and we illustrate a method for detecting fusions from single cell RNA-seq. -

Gene Signature of the Post-Chernobyl Papillary Thyroid Cancer
Eur J Nucl Med Mol Imaging (2016) 43:1267–1277 DOI 10.1007/s00259-015-3303-3 ORIGINAL ARTICLE Gene signature of the post-Chernobyl papillary thyroid cancer Daria Handkiewicz-Junak1 & Michal Swierniak1,2 & Dagmara Rusinek1 & Małgorzata Oczko-Wojciechowska1 & Genevieve Dom 3 & Carine Maenhaut3 & Kristian Unger4,5 & Vincent Detours 3 & Tetiana Bogdanova 6 & Geraldine Thomas4 & Ilya Likhtarov7 & Roman Jaksik8 & Malgorzata Kowalska1 & Ewa Chmielik9 & Michal Jarzab10 & Andrzej Swierniak11 & Barbara Jarzab1 Received: 11 August 2015 /Accepted: 29 December 2015 /Published online: 26 January 2016 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Human Genome U133 2.0 Plus) with the aim of identifying Purpose Following the nuclear accidents in Chernobyl and molecular differences between radiation-induced (exposed to later in Fukushima, the nuclear community has been faced Chernobyl radiation, ECR) and sporadic PTC. All participants with important issues concerning how to search for and diag- were resident in the same region so that confounding factors nose biological consequences of low-dose internal radiation related to genetics or environment were minimized. contamination. Although after the Chernobyl accident an in- Results There were small but significant differences in the crease in childhood papillary thyroid cancer (PTC) was ob- gene expression profiles between ECR and non-ECR PTC served, it is still not clear whether the molecular biology of (global test, p < 0.01), with 300 differently expressed probe PTCs associated with low-dose radiation exposure differs sets (p < 0.001) corresponding to 239 genes. Multifactorial from that of sporadic PTC. analysis of variance showed that besides radiation exposure Methods We investigated tissue samples from 65 children/ history, the BRAF mutation exhibited independent effects on young adults with PTC using DNA microarray (Affymetrix, the PTC expression profile; the histological subset and patient Daria Handkiewicz-Junak and Michal Swierniak contributed equally to this work. -

Cytotoxic Fractions from Hechtia Glomerata Extracts and P-Coumaric Acid As MAPK Inhibitors
molecules Article Cytotoxic Fractions from Hechtia glomerata Extracts and p-Coumaric Acid as MAPK Inhibitors Tommaso Stefani 1,* , Antonio Romo-Mancillas 2 , Juan J. J. Carrizales-Castillo 1, Eder Arredondo-Espinoza 1 , Karla Ramírez-Estrada 1 , Victor M. Alcantar-Rosales 3 , Leticia González-Maya 4 , Jessica Nayelli Sánchez-Carranza 4, Isaías Balderas-Renterías 1 and María del Rayo Camacho-Corona 1,* 1 Facultad de Ciencias Químicas, Universidad Autónoma de Nuevo León, Av. Universidad S/N, Ciudad Universitaria, San Nicolás de los Garza, NL 66451, Mexico; [email protected] (J.J.J.C.-C.); [email protected] (E.A.-E.); [email protected] (K.R.-E.); [email protected] (I.B.-R.) 2 Laboratorio de Diseño Asistido por Computadora y Síntesis de Fármacos, Facultad de Quimica, Universidad Autónoma de Querétaro, Centro Universitario, Cerro de las Campanas S/N, Santiago de Querétaro, QT 76010, Mexico; [email protected] 3 Centro de Investigación y Asistencia en Tecnología y Diseño del Estado de Jalisco, A.C. Servicios Analíticos, Sede Noreste, Parque de Investigación e Innovación Tecnológica, Vía de la Innovación 404, Apodaca, NL 66628, Mexico; [email protected] 4 Facultad de Farmacia, Universidad Autónoma del Estado de Morelos, Av. Universidad 1001, Col. Chamilpa, Cuernavaca, MO 62209, Mexico; [email protected] (L.G.-M.); [email protected] (J.N.S.-C.) Citation: Stefani, T.; Romo-Mancillas, * Correspondence: [email protected] (T.S.); [email protected] (M.d.R.C.-C.); A.; Carrizales-Castillo, J.J.J.; Arredondo- Tel.: +52-8183294000 (ext. 3414) (M.d.R.C.-C.); Fax: +52-8183529025 (M.d.R.C.-C.) Espinoza, E.; Ramírez-Estrada, K.; Alcantar-Rosales, V.M.; González- Abstract: Preliminary bioassay-guided fractionation was performed to identify cytotoxic compounds Maya, L.; Sánchez-Carranza, J.N.; from Hechtia glomerata, a plant that is used in Mexican ethnomedicine. -

The Biological Age of a Bloodstain Donor Author(S): Jack Ballantyne, Ph.D
The author(s) shown below used Federal funding provided by the U.S. Department of Justice to prepare the following resource: Document Title: The Biological Age of a Bloodstain Donor Author(s): Jack Ballantyne, Ph.D. Document Number: 251894 Date Received: July 2018 Award Number: 2009-DN-BX-K179 This resource has not been published by the U.S. Department of Justice. This resource is being made publically available through the Office of Justice Programs’ National Criminal Justice Reference Service. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. National Center for Forensic Science University of Central Florida P.O. Box 162367 · Orlando, FL 32826 Phone: 407.823.4041 Fax: 407.823.4042 Web site: http://www.ncfs.org/ Biological Evidence _________________________________________________________________________________________________________ The Biological Age of a Bloodstain Donor FINAL REPORT May 27, 2014 Department of Justice, National Institute of justice Award Number: 2009-DN-BX-K179 (1 October 2009 – 31 May 2014) _________________________________________________________________________________________________________ Principal Investigator: Jack Ballantyne, PhD Professor Department of Chemistry Associate Director for Research National Center for Forensic Science P.O. Box 162367 Orlando, FL 32816-2366 Phone: (407) 823 4440 Fax: (407) 823 4042 e-mail: [email protected] 1 This resource was prepared by the author(s) using Federal funds provided by the U.S. Department of Justice. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. -
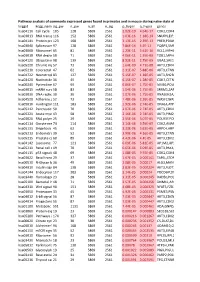
Pathway Analysis of Commonly Expressed Genes Found in Primates and in Mouse During Naïve State of Pluripotenc Keggid Kegg Names
Pathway analysis of commonly expressed genes found in primates and in mouse during naïve state of pluripotency. keggid kegg_namesig_pw n_pw n_all n_sig p_hyper q_hyper genes hsa04110 Cell cycle 105 128 5869 2561 2.02E-19 4.64E-17 CDK2,CDK4,CDK6,CDK7,CDKN1A,CDKN1B,STAG1,CDKN2B,ANAPC10,MAD2L2,STAG2,GADD45G,DBF4,YWHAQ,CHEK1,CHEK2,CREBBP,GADD45A,E2F1,E2F3,E2F4,E2F5,EP300,ORC3,CDC26,ABL1,ANAPC13,SFN,GSK3B,ANAPC2,ANAPC4,HDAC1,HDAC2,MAD2L1,SMAD2,SMAD3,SMAD4,MCM2,MCM3,MCM4,MCM5,MCM6,MCM7,MDM2,MYC,GADD45B,ATM,ORC1,ORC2,ORC4,ORC5,FZR1,ANAPC7,ANAPC11,PLK1,ATR,PRKDC,RAD21,RB1,RBL1,CCND1,ANAPC1,SKP2,BUB1,BUB1B,TFDP1,TFDP2,TGFB1,TGFB2,TTK,WEE1,YWHAB,YWHAE,YWHAG,YWHAH,YWHAZ,ZBTB17,SMC1A,CDC7,CDC45,MAD1L1,CDC14A,CDC23,CDC16,CCNA2,CCNB1,CCND2,CCND3,CCNE1,CCNH,PKMYT1,SMC3,CCNB2,CCNE2,BUB3,PTTG1,ESPL1,CDK1,CDC6,CDC20,CDC25A,CDC25B,CDC25C,CDC27,RBX1 hsa03013 RNA transport116 152 5869 2561 1.03E-16 1.18E-14 SNUPN,EIF1,POP7,SRRM1,SAP18,EIF1B,PRMT5,TACC3,NXF1,RPP30,RPP38,PAIP1,POP4,RPP40,RNPS1,POP1,STRAP,DDX20,XPOT,CLNS1A,NUP35,RPP25L,EEF1A1,EEF1A2,EIF2S1,EIF2B1,EIF4A1,EIF4B,EIF4E,EIF4EBP1,EIF4G1,EIF4G2,EIF5,CASC3,NCBP2,ACIN1,NUP205,NUP210,NUP62,GEMIN5,UPF2,PABPC1,NXT1,NUP43,EIF3E,KPNB1,MAGOH,NCBP1,NUP88,NUP98,GEMIN4,NMD3,POP5,PHAX,NUP54,PNN,RPP25,GEMIN8,MAGOHB,ELAC1,NDC1,NUP133,NXT2,NUP107,THOC2,XPO5,RAN,RANBP2,RANGAP1,SENP2,UPF1,ELAC2,SEC13,UPF3B,SMN1,SUMO3,SUMO2,TPR,SUMO1,XPO1,NUP37,DDX39B,THOC6,GEMIN7,GEMIN6,RPP21,NUP85,THOC7,NUP214,AAAS,SEH1L,THOC3,RAE1,THOC5,EIF3A,EIF3B,EIF3C,EIF3D,EIF3F,EIF3G,EIF3H,EIF3I,EIF4G3,PABPC4,EIF2B4,EIF2B2,EIF2B5,EIF2S2,EIF4E2,NUP155,EIF5B,TGS1,NUP93,EIF4A3,NUP153,THOC1 -
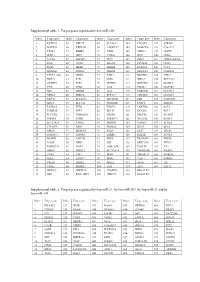
Target Genes Regulated by Hsa-Mir-21, by Hsa-Mir-203, by Hsa-Mir-21 and by Hsa-Mir-143
Supplemental table 1: Target genes regulated by hsa-miR-205 Index Target gene Index Target gene Index Target gene Index Target gene Index Target gene 1 KCTD20 35 UBE2Z 69 SLC38A1 103 LPCAT1 137 STK38L 2 MAPK14 36 YWHAH 70 ANGPTL7 104 MARCKS 138 C1orf123 3 TXNL1 37 RBBP4 71 CTGF 105 MED13 139 GUCD1 4 SPDL1 38 LRP1 72 CYR61 106 IPO7 140 CDK6 5 TCF20 39 IMPAD1 73 TP73 107 PHC2 141 CDKN2AIPNL 6 RAN 40 GNAS 74 EGLN2 108 PICALM 142 CLIP1 7 RGS6 41 MED1 75 ERBB2 109 PLAGL2 143 CUL5 8 HOXA11 42 INPPL1 76 PRRG4 110 NDUFA4 144 C6orf201 9 PAPPA-AS1 43 DDX5 77 F2RL2 111 NDUFB2 145 VTI1A 10 PRR15 44 E2F1 78 GOT1 112 NIPA2 146 SLC5A12 11 ACTRT3 45 E2F5 79 NUFIP2 113 NOTCH2 147 MAML2 12 YES1 46 ZEB2 80 IL24 114 PANK1 148 MAP3K9 13 SRC 47 ERBB3 81 IL32 115 PARD6B 149 NUDT21 14 NPRL3 48 PRKCE 82 RNF217 116 TMEM66 150 DNAJA1 15 NFAT5 49 SLC41A1 83 ZNF585B 117 EZR 151 CCDC108 16 XPOT 50 SLC7A2 84 SIGMAR1 118 ENPP4 152 SHISA6 17 KCTD16 51 ZEB1 85 VEGFA 119 LRRTM4 153 ACP1 18 TMSB4X 52 PHF8 86 BCL9L 120 KCNJ10 154 BCL2 19 PLCXD2 53 TMEM201 87 CREB1 121 PHLPP2 155 NCAPG 20 TNFSF8 54 PTPRJ 88 SERINC3 122 YEATS2 156 KLHL5 21 SLC25A25 55 ETNK1 89 HMGB3 123 VAMP1 157 ACSL4 22 C11orf74 56 XPR1 90 SRD5A1 124 RTN3 158 BCL6 23 GM2A 57 MRPL44 91 PTEN 125 RFX7 159 ITGA5 24 SMNDC1 58 TM9SF2 92 ESRRG 126 RAP2B 160 ACSL1 25 BAMBI 59 PAIP2B 93 PRLR 127 TRAF3IP1 161 EID2B 26 LCOR 60 NEK9 94 ICK 128 SERTAD2 162 TEX35 27 TMEM239 61 NOX5 95 LOH12CR1 129 TOLLIP 163 YY1 28 AMOT 62 DMXL2 96 SLC39A14 130 TMEM55B 164 SMAD1 29 CDK1 63 ETF1 97 BDP1 131 TMEM123 165 SMAD4 30 SQLE 64