Conserved Redox-Control of Ser/Thr Protein Kinase Activity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

The Role of P21-Activated Protein Kinase 1 in Metabolic Homeostasis
THE ROLE OF P21-ACTIVATED PROTEIN KINASE 1 IN METABOLIC HOMEOSTASIS by YU-TING CHIANG A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Graduate Department of Physiology University of Toronto © Copyright by Yu-ting Chiang 2014 The Role of P21-Activated Protein Kinase 1 in Metabolic Homeostasis Yu-ting Chiang Doctor of Philosophy Department of Physiology University of Toronto 2014 Abstract Our laboratory has demonstrated previously that the proglucagon gene (gcg), which encodes the incretin hormone GLP-1, is among the downstream targets of the Wnt signaling pathway; and that Pak1 mediates the stimulatory effect of insulin on Wnt target gene expression in mouse gut non- endocrine cells. Here, I asked whether Pak1 controls gut gcg expression and GLP-1 production, and whether Pak1 deletion leads to impaired metabolic homeostasis in mice. I detected the expression of Pak1 and two other group I Paks in the gut endocrine L cell line GLUTag, and co- localized Pak1 and GLP-1 in the mouse gut. Insulin was shown to stimulate Pak1 Thr423 and β-cat Ser675 phosphorylation. The stimulation of insulin on β-cat Ser675 phosphorylation, gcg promoter activity and gcg mRNA expression could be attenuated by the Pak inhibitor IPA3. Male Pak1-/- mice showed significant reduction in both gut and brain gcg expression levels, and attenuated elevation of plasma GLP-1 levels in response to oral glucose challenge. Notably, the Pak1-/- mice were intolerant to both intraperitoneal and oral glucose administration. Aged Pak1-/- mice showed a severe defect in response to intraperitoneal pyruvate challenge (IPPTT). -

MAP2K3 (Human) Recombinant Protein (Q01)
MAP2K3 (Human) Recombinant phosphorylates and thus activates MAPK14/p38-MAPK. Protein (Q01) This kinase can be activated by insulin, and is necessary for the expression of glucose transporter. Expression of Catalog Number: H00005606-Q01 RAS oncogene is found to result in the accumulation of the active form of this kinase, which thus leads to the Regulation Status: For research use only (RUO) constitutive activation of MAPK14, and confers oncogenic transformation of primary cells. The inhibition Product Description: Human MAP2K3 partial ORF ( of this kinase is involved in the pathogenesis of Yersina AAH32478, 1 a.a. - 100 a.a.) recombinant protein with pseudotuberculosis. Multiple alternatively spliced GST-tag at N-terminal. transcript variants that encode distinct isoforms have been reported for this gene. [provided by RefSeq] Sequence: MESPASSQPASMPQSKGKSKRKKDLRISCMSKPPAP NPTPPRNLDSRTFITIGDRNFEVEADDLVTISELGRGAY GVVEKVRHAQSGTIMAVKRIRATVN Host: Wheat Germ (in vitro) Theoretical MW (kDa): 36.63 Applications: AP, Array, ELISA, WB-Re (See our web site product page for detailed applications information) Protocols: See our web site at http://www.abnova.com/support/protocols.asp or product page for detailed protocols Preparation Method: in vitro wheat germ expression system Purification: Glutathione Sepharose 4 Fast Flow Storage Buffer: 50 mM Tris-HCI, 10 mM reduced Glutathione, pH=8.0 in the elution buffer. Storage Instruction: Store at -80°C. Aliquot to avoid repeated freezing and thawing. Entrez GeneID: 5606 Gene Symbol: MAP2K3 Gene Alias: MAPKK3, MEK3, MKK3, PRKMK3 Gene Summary: The protein encoded by this gene is a dual specificity protein kinase that belongs to the MAP kinase kinase family. This kinase is activated by mitogenic and environmental stress, and participates in the MAP kinase-mediated signaling cascade. -

Epithelial Ovarian Cancer: Searching for New Modulators of Drug Resistance
UNIVERSITÀ DEGLI STUDI DI UDINE Dipartimento di scienze mediche e biologiche PhD COURSE IN BIOMEDICAL SCIENCES AND BIOTECHNOLOGY XXIX cycle PhD THESIS Epithelial Ovarian Cancer: searching for new modulators of drug resistance. PhD student Supervisor Prof. Carlo Ennio Michele Pucillo Valentina Ranzuglia Co-supervisors Dr. Gustavo Baldassarre Dr. Monica Schiappacassi PhD coordinator Prof. Claudio Brancolini Academic year 2015/2016 This PhD work was done at Centro di Riferimento Oncologico (CRO, National Cancer Institute ) of Aviano, in the Division of Experimental Oncology 2 directed by Dr. Gustavo Baldassarre. i Table of contents Abstract 3 1. Introduction 4 1.1 Epithelial Ovarian Cancer 4 1.1.1 Platinum resistance 5 1.2 Functional Genomic Screening 8 1.3 Serum and glucocorticoid-regulated kinases 9 1.3.1 SGK structure 10 1.3.2 SGK activation and regulation 11 1.3.3 Role of SGKs in regulation of molecular and cellular functions 13 1.3.4 Serum- and glucocorticoid-regulated kinases in cancer 15 1.4 Autophagy 17 2.Aim of the study 21 3.Results 22 3.1 Identification of SGK2 as a mediator of platinum sensitivity in EOC cells. 22 3.2 SGK2 silencing sensitizes EOC cells to platinum. 23 3.3 SGK2 overexpression confers an increased resistance to platinum drug. 25 3.4 SGK2-overexpressing cells present an increased in vitro and in vivo growth 26 rate 3.5 SGK2 kinase activity is involved in EOC sensitivity to platinum treatment. 28 3.6 The SGK1/SGK2 kinase inhibitor, GSK650394, reduces cell viability in 29 combination with platinum. 3.7 GSK650394 is able to make SGK2-expressing cells more sensitive to platinum. -

AGC Kinases in Mtor Signaling, in Mike Hall and Fuyuhiko Tamanoi: the Enzymes, Vol
Provided for non-commercial research and educational use only. Not for reproduction, distribution or commercial use. This chapter was originally published in the book, The Enzymes, Vol .27, published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who know you, and providing a copy to your institution’s administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at: http://www.elsevier.com/locate/permissionusematerial From: ESTELA JACINTO, AGC Kinases in mTOR Signaling, In Mike Hall and Fuyuhiko Tamanoi: The Enzymes, Vol. 27, Burlington: Academic Press, 2010, pp.101-128. ISBN: 978-0-12-381539-2, © Copyright 2010 Elsevier Inc, Academic Press. Author's personal copy 7 AGC Kinases in mTOR Signaling ESTELA JACINTO Department of Physiology and Biophysics UMDNJ-Robert Wood Johnson Medical School, Piscataway New Jersey, USA I. Abstract The mammalian target of rapamycin (mTOR), a protein kinase with homology to lipid kinases, orchestrates cellular responses to growth and stress signals. Various extracellular and intracellular inputs to mTOR are known. mTOR processes these inputs as part of two mTOR protein com- plexes, mTORC1 or mTORC2. Surprisingly, despite the many cellular functions that are linked to mTOR, there are very few direct mTOR substrates identified to date. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
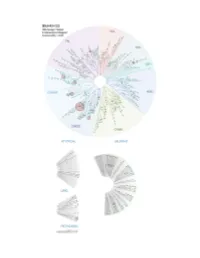
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -

Application of a MYC Degradation
SCIENCE SIGNALING | RESEARCH ARTICLE CANCER Copyright © 2019 The Authors, some rights reserved; Application of a MYC degradation screen identifies exclusive licensee American Association sensitivity to CDK9 inhibitors in KRAS-mutant for the Advancement of Science. No claim pancreatic cancer to original U.S. Devon R. Blake1, Angelina V. Vaseva2, Richard G. Hodge2, McKenzie P. Kline3, Thomas S. K. Gilbert1,4, Government Works Vikas Tyagi5, Daowei Huang5, Gabrielle C. Whiten5, Jacob E. Larson5, Xiaodong Wang2,5, Kenneth H. Pearce5, Laura E. Herring1,4, Lee M. Graves1,2,4, Stephen V. Frye2,5, Michael J. Emanuele1,2, Adrienne D. Cox1,2,6, Channing J. Der1,2* Stabilization of the MYC oncoprotein by KRAS signaling critically promotes the growth of pancreatic ductal adeno- carcinoma (PDAC). Thus, understanding how MYC protein stability is regulated may lead to effective therapies. Here, we used a previously developed, flow cytometry–based assay that screened a library of >800 protein kinase inhibitors and identified compounds that promoted either the stability or degradation of MYC in a KRAS-mutant PDAC cell line. We validated compounds that stabilized or destabilized MYC and then focused on one compound, Downloaded from UNC10112785, that induced the substantial loss of MYC protein in both two-dimensional (2D) and 3D cell cultures. We determined that this compound is a potent CDK9 inhibitor with a previously uncharacterized scaffold, caused MYC loss through both transcriptional and posttranslational mechanisms, and suppresses PDAC anchorage- dependent and anchorage-independent growth. We discovered that CDK9 enhanced MYC protein stability 62 through a previously unknown, KRAS-independent mechanism involving direct phosphorylation of MYC at Ser . -

SGK3 Sirna Set I Sirna Duplexes Targeted Against Three Exon Regions
Catalog # Aliquot Size S08-911-05 3 x 5 nmol S08-911-20 3 x 20 nmol S08-911-50 3 x 50 nmol SGK3 siRNA Set I siRNA duplexes targeted against three exon regions Catalog # S08-911 Lot # Z2085-77 Specificity Formulation SGK3 siRNAs are designed to specifically knock-down The siRNAs are supplied as a lyophilized powder and human SGK3 expression. shipped at room temperature. Product Description Reconstitution Protocol SGK3 siRNA is a pool of three individual synthetic siRNA Briefly centrifuge the tubes (maximum RCF 4,000g) to duplexes designed to knock-down human SGK3 mRNA collect lyophilized siRNA at the bottom of the tube. expression. Each siRNA is 19-25 bases in length. The gene Resuspend the siRNA in 50 µl of DEPC-treated water accession number is NM_013257. (supplied by researcher), which results in a 1x stock solution (10 µM). Gently pipet the solution 3-5 times to mix Gene Aliases and avoid the introduction of bubbles. Optional: aliquot CISK, SGKL 1x stock solutions for storage. Storage and Stability Related Products The lyophilized powder is stable for at least 4 weeks at room temperature. It is recommended that the Product Name Catalog Number lyophilized and resuspended siRNAs are stored at or SGK1, Active S06-11G below -20oC. After resuspension, siRNA stock solutions ≥2 SGK2, Active S07-10G µM can undergo up to 50 freeze-thaw cycles without SGK3, Active S08-10G significant degradation. For long-term storage, it is SGK1, Unactive S06-15G recommended that the siRNA is stored at -70oC. For most SGK2, Unactive S07-14G favorable performance, avoid repeated handling and SGK3, Unactive S08-14G multiple freeze/thaw cycles. -

Identification of DNA Response Elements Regulating Expression of CCAAT/Enhancer-Binding Protein (C/EBP) Β and Δ During Early A
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.22.110114; this version posted May 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Identification of DNA response elements regulating expression of CCAAT/enhancer-binding protein (C/EBP) β and ẟ during early adipogenesis James E. Merrett1,2, Tao Bo1,4, Peter J. Psaltis1,3 and Christopher G. Proud1,2 * 1South Australian Health and Medical Research Institute, Lifelong Health, Adelaide, South Australia, Australia 2University of Adelaide, Department of Molecular and Biomedical Science, Adelaide, South Australia, Australia 3University of Adelaide, Adelaide Medical School, Adelaide, South Australia, Australia 4Shandong-South Australia Joint Laboratory of Metabolic Disease Research, Shandong Provincial Hospital affiliated to Shandong First Medical University *Corresponding author: Christopher Proud E-mail: [email protected] Running title: DNA response elements regulating early adipogenesis Keywords: adipogenesis, adipocyte, cyclic AMP (cAMP), cAMP response element-binding protein (CREB), CCAAT/enhancer-binding protein (C/EBP), glucocorticoid, glucocorticoid receptor, MAPK- interacting kinase (MNK) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.22.110114; this version posted May 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Given the high and increasing prevalence of obesity and associated disorders, such as type-2 diabetes, it is important to understand the mechanisms that regulate lipid storage and the differentiation of fat cells, a process termed adipogenesis.