Epithelial Ovarian Cancer: Searching for New Modulators of Drug Resistance
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

AGC Kinases in Mtor Signaling, in Mike Hall and Fuyuhiko Tamanoi: the Enzymes, Vol
Provided for non-commercial research and educational use only. Not for reproduction, distribution or commercial use. This chapter was originally published in the book, The Enzymes, Vol .27, published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who know you, and providing a copy to your institution’s administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at: http://www.elsevier.com/locate/permissionusematerial From: ESTELA JACINTO, AGC Kinases in mTOR Signaling, In Mike Hall and Fuyuhiko Tamanoi: The Enzymes, Vol. 27, Burlington: Academic Press, 2010, pp.101-128. ISBN: 978-0-12-381539-2, © Copyright 2010 Elsevier Inc, Academic Press. Author's personal copy 7 AGC Kinases in mTOR Signaling ESTELA JACINTO Department of Physiology and Biophysics UMDNJ-Robert Wood Johnson Medical School, Piscataway New Jersey, USA I. Abstract The mammalian target of rapamycin (mTOR), a protein kinase with homology to lipid kinases, orchestrates cellular responses to growth and stress signals. Various extracellular and intracellular inputs to mTOR are known. mTOR processes these inputs as part of two mTOR protein com- plexes, mTORC1 or mTORC2. Surprisingly, despite the many cellular functions that are linked to mTOR, there are very few direct mTOR substrates identified to date. -
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA -

SGK3 Sirna Set I Sirna Duplexes Targeted Against Three Exon Regions
Catalog # Aliquot Size S08-911-05 3 x 5 nmol S08-911-20 3 x 20 nmol S08-911-50 3 x 50 nmol SGK3 siRNA Set I siRNA duplexes targeted against three exon regions Catalog # S08-911 Lot # Z2085-77 Specificity Formulation SGK3 siRNAs are designed to specifically knock-down The siRNAs are supplied as a lyophilized powder and human SGK3 expression. shipped at room temperature. Product Description Reconstitution Protocol SGK3 siRNA is a pool of three individual synthetic siRNA Briefly centrifuge the tubes (maximum RCF 4,000g) to duplexes designed to knock-down human SGK3 mRNA collect lyophilized siRNA at the bottom of the tube. expression. Each siRNA is 19-25 bases in length. The gene Resuspend the siRNA in 50 µl of DEPC-treated water accession number is NM_013257. (supplied by researcher), which results in a 1x stock solution (10 µM). Gently pipet the solution 3-5 times to mix Gene Aliases and avoid the introduction of bubbles. Optional: aliquot CISK, SGKL 1x stock solutions for storage. Storage and Stability Related Products The lyophilized powder is stable for at least 4 weeks at room temperature. It is recommended that the Product Name Catalog Number lyophilized and resuspended siRNAs are stored at or SGK1, Active S06-11G below -20oC. After resuspension, siRNA stock solutions ≥2 SGK2, Active S07-10G µM can undergo up to 50 freeze-thaw cycles without SGK3, Active S08-10G significant degradation. For long-term storage, it is SGK1, Unactive S06-15G recommended that the siRNA is stored at -70oC. For most SGK2, Unactive S07-14G favorable performance, avoid repeated handling and SGK3, Unactive S08-14G multiple freeze/thaw cycles. -

Kinase Profiling Book
Custom and Pre-Selected Kinase Prof iling to f it your Budget and Needs! As of July 1, 2021 19.8653 mm 128 196 12 Tyrosine Serine/Threonine Lipid Kinases Kinases Kinases Carna Biosciences, Inc. 2007 Carna Biosciences, Inc. Profiling Assays available from Carna Biosciences, Inc. As of July 1, 2021 Page Kinase Name Assay Platform Page Kinase Name Assay Platform 4 ABL(ABL1) MSA 21 EGFR[T790M/C797S/L858R] MSA 4 ABL(ABL1)[E255K] MSA 21 EGFR[T790M/L858R] MSA 4 ABL(ABL1)[T315I] MSA 21 EPHA1 MSA 4 ACK(TNK2) MSA 21 EPHA2 MSA 4 AKT1 MSA 21 EPHA3 MSA 5 AKT2 MSA 22 EPHA4 MSA 5 AKT3 MSA 22 EPHA5 MSA 5 ALK MSA 22 EPHA6 MSA 5 ALK[C1156Y] MSA 22 EPHA7 MSA 5 ALK[F1174L] MSA 22 EPHA8 MSA 6 ALK[G1202R] MSA 23 EPHB1 MSA 6 ALK[G1269A] MSA 23 EPHB2 MSA 6 ALK[L1196M] MSA 23 EPHB3 MSA 6 ALK[R1275Q] MSA 23 EPHB4 MSA 6 ALK[T1151_L1152insT] MSA 23 Erk1(MAPK3) MSA 7 EML4-ALK MSA 24 Erk2(MAPK1) MSA 7 NPM1-ALK MSA 24 Erk5(MAPK7) MSA 7 AMPKα1/β1/γ1(PRKAA1/B1/G1) MSA 24 FAK(PTK2) MSA 7 AMPKα2/β1/γ1(PRKAA2/B1/G1) MSA 24 FER MSA 7 ARG(ABL2) MSA 24 FES MSA 8 AurA(AURKA) MSA 25 FGFR1 MSA 8 AurA(AURKA)/TPX2 MSA 25 FGFR1[V561M] MSA 8 AurB(AURKB)/INCENP MSA 25 FGFR2 MSA 8 AurC(AURKC) MSA 25 FGFR2[V564I] MSA 8 AXL MSA 25 FGFR3 MSA 9 BLK MSA 26 FGFR3[K650E] MSA 9 BMX MSA 26 FGFR3[K650M] MSA 9 BRK(PTK6) MSA 26 FGFR3[V555L] MSA 9 BRSK1 MSA 26 FGFR3[V555M] MSA 9 BRSK2 MSA 26 FGFR4 MSA 10 BTK MSA 27 FGFR4[N535K] MSA 10 BTK[C481S] MSA 27 FGFR4[V550E] MSA 10 BUB1/BUB3 MSA 27 FGFR4[V550L] MSA 10 CaMK1α(CAMK1) MSA 27 FGR MSA 10 CaMK1δ(CAMK1D) MSA 27 FLT1 MSA 11 CaMK2α(CAMK2A) MSA 28 -

Supplementary Table 1. in Vitro Side Effect Profiling Study for LDN/OSU-0212320. Neurotransmitter Related Steroids
Supplementary Table 1. In vitro side effect profiling study for LDN/OSU-0212320. Percent Inhibition Receptor 10 µM Neurotransmitter Related Adenosine, Non-selective 7.29% Adrenergic, Alpha 1, Non-selective 24.98% Adrenergic, Alpha 2, Non-selective 27.18% Adrenergic, Beta, Non-selective -20.94% Dopamine Transporter 8.69% Dopamine, D1 (h) 8.48% Dopamine, D2s (h) 4.06% GABA A, Agonist Site -16.15% GABA A, BDZ, alpha 1 site 12.73% GABA-B 13.60% Glutamate, AMPA Site (Ionotropic) 12.06% Glutamate, Kainate Site (Ionotropic) -1.03% Glutamate, NMDA Agonist Site (Ionotropic) 0.12% Glutamate, NMDA, Glycine (Stry-insens Site) 9.84% (Ionotropic) Glycine, Strychnine-sensitive 0.99% Histamine, H1 -5.54% Histamine, H2 16.54% Histamine, H3 4.80% Melatonin, Non-selective -5.54% Muscarinic, M1 (hr) -1.88% Muscarinic, M2 (h) 0.82% Muscarinic, Non-selective, Central 29.04% Muscarinic, Non-selective, Peripheral 0.29% Nicotinic, Neuronal (-BnTx insensitive) 7.85% Norepinephrine Transporter 2.87% Opioid, Non-selective -0.09% Opioid, Orphanin, ORL1 (h) 11.55% Serotonin Transporter -3.02% Serotonin, Non-selective 26.33% Sigma, Non-Selective 10.19% Steroids Estrogen 11.16% 1 Percent Inhibition Receptor 10 µM Testosterone (cytosolic) (h) 12.50% Ion Channels Calcium Channel, Type L (Dihydropyridine Site) 43.18% Calcium Channel, Type N 4.15% Potassium Channel, ATP-Sensitive -4.05% Potassium Channel, Ca2+ Act., VI 17.80% Potassium Channel, I(Kr) (hERG) (h) -6.44% Sodium, Site 2 -0.39% Second Messengers Nitric Oxide, NOS (Neuronal-Binding) -17.09% Prostaglandins Leukotriene, -

An Evolutionary-Conserved Redox Regulatory Mechanism in Human Ser/Thr Protein Kinases
bioRxiv preprint doi: https://doi.org/10.1101/571844; this version posted March 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. An evolutionary-conserved redox regulatory mechanism in human Ser/Thr protein kinases Dominic P. Byrne1*, Safal Shrestha2,3, Natarajan Kannan2.3 and Patrick A. Eyers1* 1 Department of Biochemistry, Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, UK 2 Institute of Bioinformatics, University of Georgia, Athens, GA 30602, USA 3 Department of Biochemistry & Molecular Biology, University of Georgia, Athens, GA 30602, USA * Correspondence to [email protected] or [email protected] ONE-SENTENCE SUMMARY: The catalytic activity of Ser/Thr kinases is regulated through a conserved Cys-based redox mechanism. ABSTRACT: Reactive oxygen species (ROS) are products of oxygen metabolism, but are also recognized as endogenous physiological mediators of cellular signaling. Eukaryotic protein kinase (ePK) regulation occurs through reversible phosphorylation events in the flexible activation segment. In this study, we demonstrate that the catalytic phosphotransferase output from the mitotic Ser/Thr kinase Aurora A is also controlled by cysteine (Cys) oxidation. Reversible regulation occurs by direct modification of a conserved residue (Cys 290), which lies adjacent to Thr 288, the activating site of phosphorylation. Strikingly, redox modulation of the Cys 290-equivalent in other ePKs is predicted to be an underappreciated regulatory mechanism, since ~100 human Ser/Thr kinases possess a Cys at this position in the conserved activation loop. -

Combining Micro-RNA and Protein Sequencing to Detect Robust
www.nature.com/scientificreports OPEN Combining micro-RNA and protein sequencing to detect robust biomarkers for Graves’ disease and Received: 2 March 2018 Accepted: 15 May 2018 orbitopathy Published: xx xx xxxx Lei Zhang1, Giulia Masetti1,2, Giuseppe Colucci3, Mario Salvi 3, Danila Covelli3, Anja Eckstein4, Ulrike Kaiser4, Mohd Shazli Draman 1, Ilaria Muller 1, Marian Ludgate1, Luigi Lucini 5 & Filippo Biscarini 1,6 Graves’ Disease (GD) is an autoimmune condition in which thyroid-stimulating antibodies (TRAB) mimic thyroid-stimulating hormone function causing hyperthyroidism. 5% of GD patients develop infammatory Graves’ orbitopathy (GO) characterized by proptosis and attendant sight problems. A major challenge is to identify which GD patients are most likely to develop GO and has relied on TRAB measurement. We screened sera/plasma from 14 GD, 19 GO and 13 healthy controls using high-throughput proteomics and miRNA sequencing (Illumina’s HiSeq2000 and Agilent-6550 Funnel quadrupole-time-of-fight mass spectrometry) to identify potential biomarkers for diagnosis or prognosis evaluation. Euclidean distances and diferential expression (DE) based on miRNA and protein quantifcation were analysed by multidimensional scaling (MDS) and multinomial regression respectively. We detected 3025 miRNAs and 1886 proteins and MDS revealed good separation of the 3 groups. Biomarkers were identifed by combined DE and Lasso-penalized predictive models; accuracy of predictions was 0.86 (±0:18), and 5 miRNA and 20 proteins were found including Zonulin, Alpha-2 macroglobulin, Beta-2 glycoprotein 1 and Fibronectin. Functional analysis identifed relevant metabolic pathways, including hippo signaling, bacterial invasion of epithelial cells and mRNA surveillance. Proteomic and miRNA analyses, combined with robust bioinformatics, identifed circulating biomarkers applicable to diagnose GD, predict GO disease status and optimize patient management. -

SGK2, Active Full-Length Recombinant Protein Expressed in Sf9 Cells
570-5600 Parkwood Way Richmond, BC V6V2M2 CANADA Tel: 1-(604)-232-4600 Fax: 1-(604)-232-4601 [email protected] SGK2, Active Full-length recombinant protein expressed in Sf9 cells Catalog # S07-10G Lot # C162-2 Product Description Specific Activity Recombinant full-length human SGK2 was expressed by baculovirus in Sf9 insect cells using an N-terminal GST tag. 660,000 The gene accession number is NM_170693. 495,000 330,000 Gene Aliases 165,000 H-SGK2; dJ138B7.2 Activity (cpm) 0 0 80 160 240 320 Formulation Protein (ng) The specific activity of SGK2 was determined to be 94 nmol Recombinant protein stored in 50mM Tris-HCl, pH 7.5, /min/mg as per activity assay protocol. 150mM NaCl, 0.25mM DTT, 0.1mM EGTA, 0.1mM EDTA, Purity 0.1mM PMSF, 25% glycerol. Storage and Stability Store product at –70oC. For optimal storage, aliquot target into smaller quantities after centrifugation and The purity was determined to be store at recommended temperature. For most favorable >90% by densitometry. performance, avoid repeated handling and multiple Approx. MW 71kDa. freeze/thaw cycles. Scientific Background SGK2 is a member of the serum- and glucocorticoid- SGK2, Active induced kinases (SGK) which are serine-threonine kinases Full-length recombinant protein expressed in Sf9 cells and belong to the "AGC" kinase subfamily, which includes protein kinases A, G, and C, and its catalytic Catalog Number S07-10G domain is most similar to protein kinase B (PKB) (1). SGK2, Specific Activity 94 nmol/min/mg like the other two isoforms SGK1 and SGK3, is stimulated Specific Lot Number C162-2 by insulin and insulin-like growth factor-1 (IGF-1), and has Purity >90% been shown to enhance Na(+)/K(+)-ATPase activity in a Concentration 0.1µg/µl variety of cells (2). -
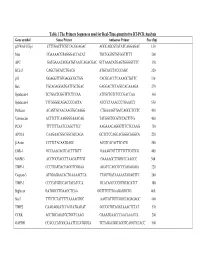
Table 1 the Primers Sequences Used for Real-Time Quantitative RT-PCR
Table 1 The Primers Sequences used for Real-Time quantitative RT-PCR Analysis Gene symbol Sense Primer Antisense Primer Size (bp) p21WAF1/Cip1 CTTTGACTTCGTCACGGAGAC AGGCAGCGTATATCAGGAGAC 150 Max CGAAAACGTAGGGACCACAT TGCTGGTGTGTGGTTTTT 200 APC GATGAAATAGGATGTAATCAGACGAC GCTAAACATGAGTGGGGTCTC 350 BCL-2 CAGCTGCACCTGACG ATGCACCTACCCAGC 320 p53 GGAGGTTGTGAGGCGCTGG CACGCACCTCAAAGCTGTTC 330 Bax TGCAGAGGATGATTGCTGAC GAGGACTCCAGCCACAAAGA 270 Syndecan-1 TCTGACTCGGTTTCTCCAA ATTGCTGTCTCCCGACCAA 360 Syndecan-3 TTTGGGGCAGACCCCACTA ATCCCTAAACCCTGAACCT 550 Perlecan ACAGTGCAACAAGTGCAAGG CTGAAAGTGACCAGGCTCCTC 450 Vitronectin ACTTCTTCAAGGGGAAACAG TATGGCTGCGTCCACTTTG 400 PCAF TTTTTTTAATCCAGCTTCC AAGAAACAGGGTTTCTCCAAG 750 APO1A CAAGAACGGCGGCGCCAGA GCTCTCCAGCACGGGCAGGCA 220 β-Actin CCTTCTACAATGAGC ACGTCACACTTCATG 260 EGR-1 GCCAAACAGTCACTTTGTT GAAAGTGTTTTTTCTTCGTCG 400 MAPK3 ACCTGCGACCTTAAGATTTGT GAAAAGCTTGGCCCAAGCC 300 TIMP-1 CCCTGATGACGAGGTCGGAA AGATCCAGCGCCCAGAGAGA 220 Caspase-3 ATGGAGAACACTGAAAACTCA TTAGTGATAAAAATAGAGTTC 200 TIMP-3 CCCCATGTGCAGTACATCCA GCACAGCCCCGTGTACATCT 180 Biglycan GATGGCCTGAAGCTCAA GGTTTGTTGAAGAGGCTG 460 Stat3 TTTCTCTATTTTTAAAAGTGC AAGTATTGTCGGCCAGAGAGC 460 TIMP2 CAAGAGGATCCAGTATGAGAT GCCCGTGTAGATAAACTCTAT 570 CCRK ACCTGCAGATGCTGCTCAAG GAAATGAACCCAACAAACCA 200 GAPDH CCACCCATGGCAAATTCCATGGGA TCTAGACGGCAGGTCAGGTCCACC 500 Table 2. Genes with Altered Expression in Cleaved vs. Native Antithrombin-Treated HUVECs Gene Fold Changes Gene Symbol Gene Description Cluster ID (ATC/ATN) Adhesion/ECM ARGN Agrin Hs.273330 2.18 CDH24 Caderhrin-like -

Serum and Glucocorticoid-Regulated Kinase 1 Regulates Neutrophil Clearance During Inflammation Resolution
Published January 15, 2014, doi:10.4049/jimmunol.1300087 The Journal of Immunology Serum and Glucocorticoid-Regulated Kinase 1 Regulates Neutrophil Clearance during Inflammation Resolution Joseph Burgon,* Anne L. Robertson,† Pranvera Sadiku,† Xingang Wang,‡ Edward Hooper-Greenhill,x Lynne R. Prince,† Paul Walker,{ Emily E. Hoggett,* Jonathan R. Ward,* Stuart N. Farrow,‖ William J. Zuercher,# Philip Jeffrey,x Caroline O. Savage,x Philip W. Ingham,‡ Adam F. Hurlstone,{ Moira K. B. Whyte,*,† and Stephen A. Renshaw*,† The inflammatory response is integral to maintaining health by functioning to resist microbial infection and repair tissue damage. Large numbers of neutrophils are recruited to inflammatory sites to neutralize invading bacteria through phagocytosis and the release of proteases and reactive oxygen species into the extracellular environment. Removal of the original inflammatory stimulus must be accompanied by resolution of the inflammatory response, including neutrophil clearance, to prevent inadvertent tissue damage. Neutrophil apoptosis and its temporary inhibition by survival signals provides a target for anti-inflammatory therapeutics, making it essential to better understand this process. GM-CSF, a neutrophil survival factor, causes a significant increase in mRNA levels for the known anti-apoptotic protein serum and glucocorticoid–regulated kinase 1 (SGK1). We have characterized the expression patterns and regulation of SGK family members in human neutrophils and shown that inhibition of SGK activity completely abrogates the antiapoptotic effect of GM-CSF. Using a transgenic zebrafish model, we have disrupted sgk1 gene function and shown this specifically delays inflammation resolution, without altering neutrophil recruitment to inflammatory sites in vivo. These data suggest SGK1 plays a key role in regulating neutrophil survival signaling and thus may prove a valuable therapeutic target for the treatment of inflammatory disease. -

Hypoxia Transcriptomic Modifications Induced by Proton Irradiation In
Journal of Personalized Medicine Article Hypoxia Transcriptomic Modifications Induced by Proton Irradiation in U87 Glioblastoma Multiforme Cell Line Valentina Bravatà 1,2,† , Walter Tinganelli 3,†, Francesco P. Cammarata 1,2,* , Luigi Minafra 1,2,* , Marco Calvaruso 1,2 , Olga Sokol 3 , Giada Petringa 2, Giuseppe A.P. Cirrone 2, Emanuele Scifoni 4 , Giusi I. Forte 1,2,‡ and Giorgio Russo 1,2,‡ 1 Institute of Molecular Bioimaging and Physiology–National Research Council (IBFM-CNR), 90015 Cefalù, Italy; [email protected] (V.B.); [email protected] (M.C.); [email protected] (G.I.F.); [email protected] (G.R.) 2 Laboratori Nazionali del SUD, Istituto Nazionale di Fisica Nucleare (LNS-INFN), 95100 Catania, Italy; [email protected] (G.P.); [email protected] (G.A.P.C.) 3 Biophysics Department, GSI Helmholtzzentrum für Schwerionenforschung GmbH, 64291 Darmstadt, Germany; [email protected] (W.T.); [email protected] (O.S.) 4 Trento Institute for Fundamental Physics and Applications (TIFPA), Istituto Nazionale Fisica Nucleare (INFN), 38123 Trento, Italy; [email protected] * Correspondence: [email protected] (F.P.C.); [email protected] (L.M.) † These authors contributed equally to this work. ‡ These authors contributed equally to this work. Abstract: In Glioblastoma Multiforme (GBM), hypoxia is associated with radioresistance and poor prognosis. Since standard GBM treatments are not always effective, new strategies are needed Citation: Bravatà, V.; Tinganelli, W.; to overcome resistance to therapeutic treatments, including radiotherapy (RT). Our study aims Cammarata, F.P.; Minafra, L.; to shed light on the biomarker network involved in a hypoxic (0.2% oxygen) GBM cell line that Calvaruso, M.; Sokol, O.; Petringa, G.; is radioresistant after proton therapy (PT). -

SGK2 Promotes Renal Cancer Progression Via Enhancing ERK 1/2 and AKT Phosphorylation
European Review for Medical and Pharmacological Sciences 2019; 23: 2756-2767 SGK2 promotes renal cancer progression via enhancing ERK 1/2 and AKT phosphorylation Y. LIU1, J.-B. CHEN2, M. ZHANG1, X.-L. ZHANG3, J.-L. MENG1, J. ZHOU1, Z.-Y. HAO1, L. ZHANG1, X.-B. ZU2, C.-Z. LIANG1 1Department of Urology, the First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China 2Department of Urology, Xiangya Hospital, Central South University, Changsha, Hunan, China 3Department of Experimental Research, Sun Yat-Sen University Cancer Center, Guangzhou, Guangdong, China Yi Liu, Jinbo Chen, and Meng Zhang contributed equally to the work Abstract. – OBJECTIVE: Increasing studies CONCLUSIONS: Our results suggested that reported that the serum- and glucocorticoid-in- SGK2 promoted RCC progression by mediat- ducible kinases (SGKs) contributed to the tum- ing the phosphorylation of extracellular regulat- origenesis of various cancer. In this article, we ed protein kinases (ERK) 1/2 and Protein kinase are aiming to explore the function of SGK2 in re- B (AKT/PKB), indicating that SGK2 might serve nal cell cancer (RCC). as a potential prognostic marker and therapeu- PATIENTS AND METHODS: In this study, tic target for renal cancer patients. the SGK2 expression was quantified by West- ern blot (WB) in multiple RCC cell lines. And Key Words in vitro SGK2 knockdown and overexpression Serum- and glucocorticoid-inducible kinases (SGKs), experiments were also performed. In addition, Renal cell cancer (RCC), Extracellular regulated protein molecular function analysis was performed us- kinases (ERK), Protein kinase B (AKT/PKB). ing FunRich software V3. The Cancer Genome Atlas (TCGA) database was retrieved to verify the association between the SGK2 expression Abbreviations and the prognosis of RCC patients.