BRAFV600E-Mutant Cancers Display a Variety of Networks by SWIM Analysis: Prediction of Vemurafenib Clinical Response
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Systematic Screening for Potential Therapeutic Targets in Osteosarcoma Through a Kinome-Wide CRISPR-Cas9 Library
Cancer Biol Med 2020. doi: 10.20892/j.issn.2095-3941.2020.0162 ORIGINAL ARTICLE Systematic screening for potential therapeutic targets in osteosarcoma through a kinome-wide CRISPR-Cas9 library Yuanzhong Wu*, Liwen Zhou*, Zifeng Wang, Xin Wang, Ruhua Zhang, Lisi Zheng, Tiebang Kang Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangzhou 510060, China ABSTRACT Objective: Osteosarcoma is the most common primary malignant bone tumor. However, the survival of patients with osteosarcoma has remained unchanged during the past 30 years, owing to a lack of efficient therapeutic targets. Methods: We constructed a kinome-targeting CRISPR-Cas9 library containing 507 kinases and 100 nontargeting controls and screened the potential kinase targets in osteosarcoma. The CRISPR screening sequencing data were analyzed with the Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) Python package. The functional data were applied in the 143B cell line through lenti-CRISPR-mediated gene knockout. The clinical significance of kinases in the survival of patients with osteosarcoma was analyzed in the R2: Genomics Analysis and Visualization Platform. Results: We identified 53 potential kinase targets in osteosarcoma. Among these targets, we analyzed 3 kinases, TRRAP, PKMYT1, and TP53RK, to validate their oncogenic functions in osteosarcoma. PKMYT1 and TP53RK showed higher expression in osteosarcoma than in normal bone tissue, whereas TRRAP showed no significant difference. High expression of all 3 kinases was associated with relatively poor prognosis in patients with osteosarcoma. Conclusions: Our results not only offer potential therapeutic kinase targets in osteosarcoma but also provide a paradigm for functional genetic screening by using a CRISPR-Cas9 library, including target design, library construction, screening workflow, data analysis, and functional validation. -

Epithelial Ovarian Cancer: Searching for New Modulators of Drug Resistance
UNIVERSITÀ DEGLI STUDI DI UDINE Dipartimento di scienze mediche e biologiche PhD COURSE IN BIOMEDICAL SCIENCES AND BIOTECHNOLOGY XXIX cycle PhD THESIS Epithelial Ovarian Cancer: searching for new modulators of drug resistance. PhD student Supervisor Prof. Carlo Ennio Michele Pucillo Valentina Ranzuglia Co-supervisors Dr. Gustavo Baldassarre Dr. Monica Schiappacassi PhD coordinator Prof. Claudio Brancolini Academic year 2015/2016 This PhD work was done at Centro di Riferimento Oncologico (CRO, National Cancer Institute ) of Aviano, in the Division of Experimental Oncology 2 directed by Dr. Gustavo Baldassarre. i Table of contents Abstract 3 1. Introduction 4 1.1 Epithelial Ovarian Cancer 4 1.1.1 Platinum resistance 5 1.2 Functional Genomic Screening 8 1.3 Serum and glucocorticoid-regulated kinases 9 1.3.1 SGK structure 10 1.3.2 SGK activation and regulation 11 1.3.3 Role of SGKs in regulation of molecular and cellular functions 13 1.3.4 Serum- and glucocorticoid-regulated kinases in cancer 15 1.4 Autophagy 17 2.Aim of the study 21 3.Results 22 3.1 Identification of SGK2 as a mediator of platinum sensitivity in EOC cells. 22 3.2 SGK2 silencing sensitizes EOC cells to platinum. 23 3.3 SGK2 overexpression confers an increased resistance to platinum drug. 25 3.4 SGK2-overexpressing cells present an increased in vitro and in vivo growth 26 rate 3.5 SGK2 kinase activity is involved in EOC sensitivity to platinum treatment. 28 3.6 The SGK1/SGK2 kinase inhibitor, GSK650394, reduces cell viability in 29 combination with platinum. 3.7 GSK650394 is able to make SGK2-expressing cells more sensitive to platinum. -

AGC Kinases in Mtor Signaling, in Mike Hall and Fuyuhiko Tamanoi: the Enzymes, Vol
Provided for non-commercial research and educational use only. Not for reproduction, distribution or commercial use. This chapter was originally published in the book, The Enzymes, Vol .27, published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who know you, and providing a copy to your institution’s administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at: http://www.elsevier.com/locate/permissionusematerial From: ESTELA JACINTO, AGC Kinases in mTOR Signaling, In Mike Hall and Fuyuhiko Tamanoi: The Enzymes, Vol. 27, Burlington: Academic Press, 2010, pp.101-128. ISBN: 978-0-12-381539-2, © Copyright 2010 Elsevier Inc, Academic Press. Author's personal copy 7 AGC Kinases in mTOR Signaling ESTELA JACINTO Department of Physiology and Biophysics UMDNJ-Robert Wood Johnson Medical School, Piscataway New Jersey, USA I. Abstract The mammalian target of rapamycin (mTOR), a protein kinase with homology to lipid kinases, orchestrates cellular responses to growth and stress signals. Various extracellular and intracellular inputs to mTOR are known. mTOR processes these inputs as part of two mTOR protein com- plexes, mTORC1 or mTORC2. Surprisingly, despite the many cellular functions that are linked to mTOR, there are very few direct mTOR substrates identified to date. -
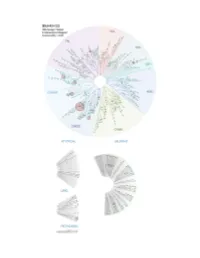
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -

Genome-Wide Sirna Screen for Mediators of NF-Κb Activation
Genome-wide siRNA screen for mediators SEE COMMENTARY of NF-κB activation Benjamin E. Gewurza, Fadi Towficb,c,1, Jessica C. Marb,d,1, Nicholas P. Shinnersa,1, Kaoru Takasakia, Bo Zhaoa, Ellen D. Cahir-McFarlanda, John Quackenbushe, Ramnik J. Xavierb,c, and Elliott Kieffa,2 aDepartment of Medicine and Microbiology and Molecular Genetics, Channing Laboratory, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115; bCenter for Computational and Integrative Biology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114; cProgram in Medical and Population Genetics, The Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02142; dDepartment of Biostatistics, Harvard School of Public Health, Boston, MA 02115; and eDepartment of Biostatistics and Computational Biology and Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA 02115 Contributed by Elliott Kieff, December 16, 2011 (sent for review October 2, 2011) Although canonical NFκB is frequently critical for cell proliferation, (RIPK1). TRADD engages TNFR-associated factor 2 (TRAF2), survival, or differentiation, NFκB hyperactivation can cause malig- which recruits the ubiquitin (Ub) E2 ligase UBC5 and the E3 nant, inflammatory, or autoimmune disorders. Despite intensive ligases cIAP1 and cIAP2. CIAP1/2 polyubiquitinate RIPK1 and study, mammalian NFκB pathway loss-of-function RNAi analyses TRAF2, which recruit and activate the K63-Ub binding proteins have been limited to specific protein classes. We therefore under- TAB1, TAB2, and TAB3, as well as their associated kinase took a human genome-wide siRNA screen for novel NFκB activa- MAP3K7 (TAK1). TAK1 in turn phosphorylates IKKβ activa- tion pathway components. Using an Epstein Barr virus latent tion loop serines to promote IKK activity (4). -

SGK3 Sirna Set I Sirna Duplexes Targeted Against Three Exon Regions
Catalog # Aliquot Size S08-911-05 3 x 5 nmol S08-911-20 3 x 20 nmol S08-911-50 3 x 50 nmol SGK3 siRNA Set I siRNA duplexes targeted against three exon regions Catalog # S08-911 Lot # Z2085-77 Specificity Formulation SGK3 siRNAs are designed to specifically knock-down The siRNAs are supplied as a lyophilized powder and human SGK3 expression. shipped at room temperature. Product Description Reconstitution Protocol SGK3 siRNA is a pool of three individual synthetic siRNA Briefly centrifuge the tubes (maximum RCF 4,000g) to duplexes designed to knock-down human SGK3 mRNA collect lyophilized siRNA at the bottom of the tube. expression. Each siRNA is 19-25 bases in length. The gene Resuspend the siRNA in 50 µl of DEPC-treated water accession number is NM_013257. (supplied by researcher), which results in a 1x stock solution (10 µM). Gently pipet the solution 3-5 times to mix Gene Aliases and avoid the introduction of bubbles. Optional: aliquot CISK, SGKL 1x stock solutions for storage. Storage and Stability Related Products The lyophilized powder is stable for at least 4 weeks at room temperature. It is recommended that the Product Name Catalog Number lyophilized and resuspended siRNAs are stored at or SGK1, Active S06-11G below -20oC. After resuspension, siRNA stock solutions ≥2 SGK2, Active S07-10G µM can undergo up to 50 freeze-thaw cycles without SGK3, Active S08-10G significant degradation. For long-term storage, it is SGK1, Unactive S06-15G recommended that the siRNA is stored at -70oC. For most SGK2, Unactive S07-14G favorable performance, avoid repeated handling and SGK3, Unactive S08-14G multiple freeze/thaw cycles. -

Resistance to Drugs and Cell Death in Cancer Stem Cells (Cscs)
Journal of Translational Science Review Article ISSN: 2059-268X Resistance to drugs and cell death in cancer stem cells (CSCs) Ahmad R Safa* Department of Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, USA Abstract Human cancers emerge from cancer stem cells (CSCs), which are resistant to cancer chemotherapeutic agents, radiation, and cell death. Moreover, autophagy provides the cytoprotective effect which contributes to drug resistance in these cells. Furthermore, much evidence shows that CSCs cause tumor initiation, progression, metastasis, and cancer recurrence. Various signaling pathways including the phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR), maternal embryonic leucine zipper kinase (MELK), NOTCH1, and Wnt/β-catenin as well as the CSC markers maintain CSC properties. Several mechanisms including overexpression of ABC multidrug resistance transporters, a deficiency in mitochondrial-mediated apoptosis, upregulation of c-FLIP, overexpression of anti- apoptotic Bcl-2 family members and inhibitors of apoptosis proteins (IAPs), and PI3K/AKT signaling contribute to enhancing resistance to chemotherapeutic drugs and cell death induction in CSCs in various cancers. Studying such pathways may help provide detailed understanding of CSC mechanisms of resistance to chemotherapeutic agents and apoptosis and may lead to the development of effective therapeutics to eradicate CSCs. Abbreviations: Apaf-1: Apoptotic Proteinase-Activating Factor-1; Tumor recurrence is the major cause of death in patients with AIC: Apoptosis Inhibitory Complex; BH3: Bcl-2 Homology Domain-3; incurable malignancies and is attributed to treatment-resistant CSCs CSCs: Cancer Stem Cells; c-FLIP: cellular FLICE (FADD-like IL- within the primary tumor (Figure 2). CSCs are a rare cell population 1β-converting enzyme)-Inhibitory Protein; DISC: Death-Inducing within a tumor, which express differing molecular markers in various Signaling Complex; DNA-PK: DNA-Dependent Protein Kinase; FADD: types of cancers [1,2,4]. -

Germline Mutations Causing Familial Lung Cancer
Journal of Human Genetics (2015) 60, 597–603 & 2015 The Japan Society of Human Genetics All rights reserved 1434-5161/15 www.nature.com/jhg ORIGINAL ARTICLE Germline mutations causing familial lung cancer Koichi Tomoshige1,2, Keitaro Matsumoto1, Tomoshi Tsuchiya1, Masahiro Oikawa1, Takuro Miyazaki1, Naoya Yamasaki1, Hiroyuki Mishima2, Akira Kinoshita2, Toru Kubo3, Kiyoyasu Fukushima3, Koh-ichiro Yoshiura2 and Takeshi Nagayasu1 Genetic factors are important in lung cancer, but as most lung cancers are sporadic, little is known about inherited genetic factors. We identified a three-generation family with suspected autosomal dominant inherited lung cancer susceptibility. Sixteen individuals in the family had lung cancer. To identify the gene(s) that cause lung cancer in this pedigree, we extracted DNA from the peripheral blood of three individuals and from the blood of one cancer-free control family member and performed whole-exome sequencing. We identified 41 alterations in 40 genes in all affected family members but not in the unaffected member. These were considered candidate mutations for familial lung cancer. Next, to identify somatic mutations and/or inherited alterations in these 40 genes among sporadic lung cancers, we performed exon target enrichment sequencing using 192 samples from sporadic lung cancer patients. We detected somatic ‘candidate’ mutations in multiple sporadic lung cancer samples; MAST1, CENPE, CACNB2 and LCT were the most promising candidate genes. In addition, the MAST1 gene was located in a putative cancer-linked locus in the pedigree. Our data suggest that several genes act as oncogenic drivers in this family, and that MAST1 is most likely to cause lung cancer. -

14-3-3 Proteins Inactivate DAPK2 by Promoting Its Dimerization And
ARTICLE https://doi.org/10.1038/s42003-021-02518-y OPEN 14-3-3 proteins inactivate DAPK2 by promoting its dimerization and protecting key regulatory phosphosites ✉ ✉ Matej Horvath 1,2, Olivia Petrvalska1,2, Petr Herman 3, Veronika Obsilova 2 & Tomas Obsil 1,2 Death-associated protein kinase 2 (DAPK2) is a CaM-regulated Ser/Thr protein kinase, involved in apoptosis, autophagy, granulocyte differentiation and motility regulation, whose activity is controlled by autoinhibition, autophosphorylation, dimerization and interaction with scaffolding proteins 14-3-3. However, the structural basis of 14-3-3-mediated DAPK2 reg- ulation remains unclear. Here, we structurally and biochemically characterize the full-length 1234567890():,; human DAPK2:14-3-3 complex by combining several biophysical techniques. The results from our X-ray crystallographic analysis revealed that Thr369 phosphorylation at the DAPK2 C terminus creates a high-affinity canonical mode III 14-3-3-binding motif, further enhanced by the diterpene glycoside Fusicoccin A. Moreover, concentration-dependent DAPK2 dimerization is disrupted by Ca2+/CaM binding and stabilized by 14-3-3 binding in solution, thereby protecting the DAPK2 inhibitory autophosphorylation site Ser318 against depho- sphorylation and preventing Ca2+/CaM binding. Overall, our findings provide mechanistic insights into 14-3-3-mediated DAPK2 inhibition and highlight the potential of the DAPK2:14- 3-3 complex as a target for anti‐inflammatory therapies. 1 Department of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Prague, Czech Republic. 2 Department of Structural Biology of Signaling Proteins, Division BIOCEV, Institute of Physiology of the Czech Academy of Sciences, Vestec, Czech Republic. 3 Institute of Physics, Faculty of ✉ Mathematics and Physics, Charles University, Prague, Czech Republic.