AGC Kinases in Mtor Signaling, in Mike Hall and Fuyuhiko Tamanoi: the Enzymes, Vol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Anti-PRKCA / PKC-Alpha Antibody (Aa623-672) Rabbit Anti Human Polyclonal Antibody Catalog # ALS17942
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Anti-PRKCA / PKC-Alpha Antibody (aa623-672) Rabbit Anti Human Polyclonal Antibody Catalog # ALS17942 Specification Anti-PRKCA / PKC-Alpha Antibody (aa623-672) - Product Information Application IHC-P, E Primary Accession P17252 Predicted Human, Mouse, Rat Host Rabbit Clonality Polyclonal Isotype IgG Calculated MW 76750 Anti-PRKCA / PKC-Alpha Antibody (aa623-672) - Additional Information Gene ID 5578 Alias Symbol PRKCA Other Names PRKCA, Aging-associated gene 6, AAG6, Alpha PKC, PKC-A, Pkcalpha, Protein kinase C alpha type, PKC Alpha, PKC-alpha, PKCA, Protein kinase c alpha, Protein kinase C, alpha Target/Specificity PRKCA Antibody detects endogenous levels of total PRKCA protein. Reconstitution & Storage Immunoaffinity purified Precautions Anti-PRKCA / PKC-Alpha Antibody (aa623-672) is for research use only and not for use in diagnostic or therapeutic procedures. Anti-PRKCA / PKC-Alpha Antibody (aa623-672) - Protein Information Name PRKCA Synonyms PKCA, PRKACA Function Page 1/4 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Calcium-activated, phospholipid- and diacylglycerol (DAG)- dependent serine/threonine-protein kinase that is involved in positive and negative regulation of cell proliferation, apoptosis, differentiation, migration and adhesion, tumorigenesis, cardiac hypertrophy, angiogenesis, platelet function and inflammation, by directly phosphorylating targets such as RAF1, BCL2, CSPG4, TNNT2/CTNT, or activating signaling cascade involving MAPK1/3 (ERK1/2) and RAP1GAP. Involved in cell proliferation and cell growth arrest by positive and negative regulation of the cell cycle. Can promote cell growth by phosphorylating and activating RAF1, which mediates the activation of the MAPK/ERK signaling cascade, and/or by up-regulating CDKN1A, which facilitates active cyclin-dependent kinase (CDK) complex formation in glioma cells. -

Generation of the Catalytic Fragment of Protein Kinase C Alpha in Vasospastic Canine Basilar Artery
Neurosurg Focus 3 (4):Article 4, 1997. Generation of the catalytic fragment of protein kinase C alpha in vasospastic canine basilar artery Motohiko Sato, M.D., Eiichi Tani, M.D., Tsuyoshi Matsumoto, M.D., Hirokazu Fujikawa, M.D., and Shinobu Imajoh-Ohmi, Ph.D. Department of Neurosurgery, Hyogo College of Medicine, Hyogo, Japan; and Institute of Medical Science, University of Tokyo, Tokyo, Japan In previous studies of topical application of calphostin C, a specific inhibitor of the regulatory domain of protein kinase C (PKC), and calpeptin, a selective inhibitor of calpain, to spastic canine basilar artery (BA) researchers have suggested that the catalytic fragment of PKC (known as PKM) is probably formed by a limited proteolysis of continuously activated µ-calpain, but there has been no direct evidence for PKM formation in vasospasm. The present immunoblot study with anti-PKC-alpha antibody shows a significant decrease in cytosolic 80-kD PKC-alpha and a concomitantly significant increase in membrane PKC-alpha in the spastic canine BA. In addition, an immunoblot study in which cleavage sitedirected antibodies were used demonstrated a significant increase in immunoreactive 45-kD PKM. The changes in membrane PKC-alpha and PKM were enhanced with the lapse of time after subarachnoid hemorrhage. The cleavage sitedirected antibodies distinguish the proteolyzed from the unproteolyzed forms of PKC for in situ analyses of enzyme regulation mediated by proteolysis. The data indicate that PKC-alpha in spastic canine BA is translocated to the cell membrane, where PKC-alpha is rapidly cleaved into PKM as a result of proteolysis of the isozyme by µ-calpain but not by m-calpain. -

ROS Production Induced by BRAF Inhibitor Treatment Rewires
Cesi et al. Molecular Cancer (2017) 16:102 DOI 10.1186/s12943-017-0667-y RESEARCH Open Access ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells Giulia Cesi, Geoffroy Walbrecq, Andreas Zimmer, Stephanie Kreis*† and Claude Haan† Abstract Background: Most melanoma patients with BRAFV600E positive tumors respond well to a combination of BRAF kinase and MEK inhibitors. However, some patients are intrinsically resistant while the majority of patients eventually develop drug resistance to the treatment. For patients insufficiently responding to BRAF and MEK inhibitors, there is an ongoing need for new treatment targets. Cellular metabolism is such a promising new target line: mutant BRAFV600E has been shown to affect the metabolism. Methods: Time course experiments and a series of western blots were performed in a panel of BRAFV600E and BRAFWT/ NRASmut human melanoma cells, which were incubated with BRAF and MEK1 kinase inhibitors. siRNA approaches were used to investigate the metabolic players involved. Reactive oxygen species (ROS) were measured by confocal microscopy and AZD7545, an inhibitor targeting PDKs (pyruvate dehydrogenase kinase) was tested. Results: We show that inhibition of the RAS/RAF/MEK/ERK pathway induces phosphorylation of the pyruvate dehydrogenase PDH-E1α subunit in BRAFV600E and in BRAFWT/NRASmut harboring cells. Inhibition of BRAF, MEK1 and siRNA knock-down of ERK1/2 mediated phosphorylation of PDH. siRNA-mediated knock-down of all PDKs or the use of DCA (a pan-PDK inhibitor) abolished PDH-E1α phosphorylation. BRAF inhibitor treatment also induced the upregulation of ROS, concomitantly with the induction of PDH phosphorylation. -

PKC Alpha Rabbit Pab
Leader in Biomolecular Solutions for Life Science PKC alpha Rabbit pAb Catalog No.: A13342 Basic Information Background Catalog No. Protein kinase C (PKC) is a family of serine- and threonine-specific protein kinases that A13342 can be activated by calcium and the second messenger diacylglycerol. PKC family members phosphorylate a wide variety of protein targets and are known to be involved Observed MW in diverse cellular signaling pathways. PKC family members also serve as major 85kDa receptors for phorbol esters, a class of tumor promoters. Each member of the PKC family has a specific expression profile and is believed to play a distinct role in cells. The Calculated MW protein encoded by this gene is one of the PKC family members. This kinase has been 76kDa reported to play roles in many different cellular processes, such as cell adhesion, cell transformation, cell cycle checkpoint, and cell volume control. Knockout studies in mice Category suggest that this kinase may be a fundamental regulator of cardiac contractility and Ca(2+) handling in myocytes. Primary antibody Applications WB Cross-Reactivity Human, Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 5578 P17252 Immunogen Recombinant fusion protein containing a sequence corresponding to amino acids 523-672 of human PKC alpha (NP_002728.1). Synonyms PRKCA;AAG6;PKC-alpha;PKCA;PRKACA;PKC alpha Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts of various cell lines, using PKC alpha antibody (A13342) at 1:1000 dilution. -

The Role of Z-Disc Proteins in Myopathy and Cardiomyopathy
International Journal of Molecular Sciences Review The Role of Z-disc Proteins in Myopathy and Cardiomyopathy Kirsty Wadmore 1,†, Amar J. Azad 1,† and Katja Gehmlich 1,2,* 1 Institute of Cardiovascular Sciences, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT, UK; [email protected] (K.W.); [email protected] (A.J.A.) 2 Division of Cardiovascular Medicine, Radcliffe Department of Medicine and British Heart Foundation Centre of Research Excellence Oxford, University of Oxford, Oxford OX3 9DU, UK * Correspondence: [email protected]; Tel.: +44-121-414-8259 † These authors contributed equally. Abstract: The Z-disc acts as a protein-rich structure to tether thin filament in the contractile units, the sarcomeres, of striated muscle cells. Proteins found in the Z-disc are integral for maintaining the architecture of the sarcomere. They also enable it to function as a (bio-mechanical) signalling hub. Numerous proteins interact in the Z-disc to facilitate force transduction and intracellular signalling in both cardiac and skeletal muscle. This review will focus on six key Z-disc proteins: α-actinin 2, filamin C, myopalladin, myotilin, telethonin and Z-disc alternatively spliced PDZ-motif (ZASP), which have all been linked to myopathies and cardiomyopathies. We will summarise pathogenic variants identified in the six genes coding for these proteins and look at their involvement in myopathy and cardiomyopathy. Listing the Minor Allele Frequency (MAF) of these variants in the Genome Aggregation Database (GnomAD) version 3.1 will help to critically re-evaluate pathogenicity based on variant frequency in normal population cohorts. -

Epithelial Ovarian Cancer: Searching for New Modulators of Drug Resistance
UNIVERSITÀ DEGLI STUDI DI UDINE Dipartimento di scienze mediche e biologiche PhD COURSE IN BIOMEDICAL SCIENCES AND BIOTECHNOLOGY XXIX cycle PhD THESIS Epithelial Ovarian Cancer: searching for new modulators of drug resistance. PhD student Supervisor Prof. Carlo Ennio Michele Pucillo Valentina Ranzuglia Co-supervisors Dr. Gustavo Baldassarre Dr. Monica Schiappacassi PhD coordinator Prof. Claudio Brancolini Academic year 2015/2016 This PhD work was done at Centro di Riferimento Oncologico (CRO, National Cancer Institute ) of Aviano, in the Division of Experimental Oncology 2 directed by Dr. Gustavo Baldassarre. i Table of contents Abstract 3 1. Introduction 4 1.1 Epithelial Ovarian Cancer 4 1.1.1 Platinum resistance 5 1.2 Functional Genomic Screening 8 1.3 Serum and glucocorticoid-regulated kinases 9 1.3.1 SGK structure 10 1.3.2 SGK activation and regulation 11 1.3.3 Role of SGKs in regulation of molecular and cellular functions 13 1.3.4 Serum- and glucocorticoid-regulated kinases in cancer 15 1.4 Autophagy 17 2.Aim of the study 21 3.Results 22 3.1 Identification of SGK2 as a mediator of platinum sensitivity in EOC cells. 22 3.2 SGK2 silencing sensitizes EOC cells to platinum. 23 3.3 SGK2 overexpression confers an increased resistance to platinum drug. 25 3.4 SGK2-overexpressing cells present an increased in vitro and in vivo growth 26 rate 3.5 SGK2 kinase activity is involved in EOC sensitivity to platinum treatment. 28 3.6 The SGK1/SGK2 kinase inhibitor, GSK650394, reduces cell viability in 29 combination with platinum. 3.7 GSK650394 is able to make SGK2-expressing cells more sensitive to platinum. -

PDK1 Acquires PDK2 Activity in the Presence of a Synthetic Peptide
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Research Paper 393 PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2 Anudharan Balendran*†, Antonio Casamayor*†, Maria Deak†, Andrew Paterson†, Piers Gaffney‡, Richard Currie§, C. Peter Downes§ and Dario R. Alessi† Background: Protein kinase B (PKB) is activated by phosphorylation of Thr308 Addresses: *MRC Protein Phosphorylation Unit, and of Ser473. Thr308 is phosphorylated by the 3-phosphoinositide-dependent Department of Biochemistry, University of Dundee, ‡ protein kinase-1 (PDK1) but the identity of the kinase that phosphorylates Dundee DD1 5EH, UK. Ludwig Institute of Cancer Research, London W1P 8BY, UK. §Department of Ser473 (provisionally termed PDK2) is unknown. Biochemistry, University of Dundee, Dundee DD1 5EH, UK. Results: The kinase domain of PDK1 interacts with a region of protein kinase C-related kinase-2 (PRK2), termed the PDK1-interacting fragment (PIF). PIF is Correspondence: Dario R. Alessi E-mail: [email protected] situated carboxy-terminal to the kinase domain of PRK2, and contains a consensus motif for phosphorylation by PDK2 similar to that found in PKBα, †A.B. and A.C. contributed equally to this work. except that the residue equivalent to Ser473 is aspartic acid. Mutation of any of the conserved residues in the PDK2 motif of PIF prevented interaction of PIF Received: 12 January 1999 Revised: 18 February 1999 with PDK1. Remarkably, interaction of PDK1 with PIF, or with a synthetic Accepted: 3 March 1999 peptide encompassing the PDK2 consensus sequence of PIF, converted PDK1 from an enzyme that could phosphorylate only Thr308 of PKBα to one that Published: 8 April 1999 phosphorylates both Thr308 and Ser473 of PKBα in a manner dependent on Current Biology 1999, 9:393–404 phosphatidylinositol (3,4,5) trisphosphate (PtdIns(3,4,5)P3). -

Phosphatidylinositol-3-Kinase Related Kinases (Pikks) in Radiation-Induced Dna Damage
Mil. Med. Sci. Lett. (Voj. Zdrav. Listy) 2012, vol. 81(4), p. 177-187 ISSN 0372-7025 DOI: 10.31482/mmsl.2012.025 REVIEW ARTICLE PHOSPHATIDYLINOSITOL-3-KINASE RELATED KINASES (PIKKS) IN RADIATION-INDUCED DNA DAMAGE Ales Tichy 1, Kamila Durisova 1, Eva Novotna 1, Lenka Zarybnicka 1, Jirina Vavrova 1, Jaroslav Pejchal 2, Zuzana Sinkorova 1 1 Department of Radiobiology, Faculty of Health Sciences in Hradec Králové, University of Defence in Brno, Czech Republic 2 Centrum of Advanced Studies, Faculty of Health Sciences in Hradec Králové, University of Defence in Brno, Czech Republic. Received 5 th September 2012. Revised 27 th November 2012. Published 7 th December 2012. Summary This review describes a drug target for cancer therapy, family of phosphatidylinositol-3 kinase related kinases (PIKKs), and it gives a comprehensive review of recent information. Besides general information about phosphatidylinositol-3 kinase superfamily, it characterizes a DNA-damage response pathway since it is monitored by PIKKs. Key words: PIKKs; ATM; ATR; DNA-PK; Ionising radiation; DNA-repair ABBREVIATIONS therapy and radiation play a pivotal role. Since cancer is one of the leading causes of death worldwide, it is DSB - double stand breaks, reasonable to invest time and resources in the enligh - IR - ionising radiation, tening of mechanisms, which underlie radio-resis - p53 - TP53 tumour suppressors, tance. PI - phosphatidylinositol. The aim of this review is to describe the family INTRODUCTION of phosphatidyinositol 3-kinases (PI3K) and its func - tional subgroup - phosphatidylinositol-3-kinase rela - An efficient cancer treatment means to restore ted kinases (PIKKs) and their relation to repairing of controlled tissue growth via interfering with cell sig - radiation-induced DNA damage. -

A Calcium- and Calmodulin-Dependent Kinase I␣/ Microtubule Affinity Regulating Kinase 2 Signaling Cascade Mediates Calcium-Dependent Neurite Outgrowth
The Journal of Neuroscience, April 18, 2007 • 27(16):4413–4423 • 4413 Cellular/Molecular A Calcium- and Calmodulin-Dependent Kinase I␣/ Microtubule Affinity Regulating Kinase 2 Signaling Cascade Mediates Calcium-Dependent Neurite Outgrowth Nataliya V. Uboha,1 Marc Flajolet,2 Angus C. Nairn,1,2 and Marina R. Picciotto1 1Department of Psychiatry, Yale University School of Medicine, New Haven, Connecticut 06508, and 2Laboratory of Molecular and Cellular Neuroscience, The Rockefeller University, New York, New York 10021 Calcium is a critical regulator of neuronal differentiation and neurite outgrowth during development, as well as synaptic plasticity in adulthood. Calcium- and calmodulin-dependent kinase I (CaMKI) can regulate neurite outgrowth; however, the signal transduction cascades that lead to its physiological effects have not yet been elucidated. CaMKI␣ was therefore used as bait in a yeast two-hybrid assay and microtubule affinity regulating kinase 2 (MARK2)/Par-1b was identified as an interacting partner of CaMKI in three independent screens. The interaction between CaMKI and MARK2 was confirmed in vitro and in vivo by coimmunoprecipitation. CaMKI binds MARK2 within its kinase domain, but only if it is activated by calcium and calmodulin. Expression of CaMKI and MARK2 in Neuro-2A (N2a) cells and in primary hippocampal neurons promotes neurite outgrowth, an effect dependent on the catalytic activities of these enzymes. In addition, decreasing MARK2 activity blocks the ability of the calcium ionophore ionomycin to promote neurite outgrowth. Finally, CaMKI phosphorylates MARK2 on novel sites within its kinase domain. Mutation of these phosphorylation sites decreases both MARK2 kinase activity and its ability to promote neurite outgrowth. -
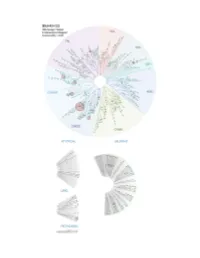
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA