The P90 RSK Family Members: Common Functions and Isoform Specificity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Aberrant Modulation of Ribosomal Protein S6 Phosphorylation Confers Acquired Resistance to MAPK Pathway Inhibitors in BRAF-Mutant Melanoma
www.nature.com/aps ARTICLE Aberrant modulation of ribosomal protein S6 phosphorylation confers acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma Ming-zhao Gao1,2, Hong-bin Wang1,2, Xiang-ling Chen1,2, Wen-ting Cao1,LiFu1, Yun Li1, Hai-tian Quan1,2, Cheng-ying Xie1,2 and Li-guang Lou1,2 BRAF and MEK inhibitors have shown remarkable clinical efficacy in BRAF-mutant melanoma; however, most patients develop resistance, which limits the clinical benefit of these agents. In this study, we found that the human melanoma cell clones, A375-DR and A375-TR, with acquired resistance to BRAF inhibitor dabrafenib and MEK inhibitor trametinib, were cross resistant to other MAPK pathway inhibitors. In these resistant cells, phosphorylation of ribosomal protein S6 (rpS6) but not phosphorylation of ERK or p90 ribosomal S6 kinase (RSK) were unable to be inhibited by MAPK pathway inhibitors. Notably, knockdown of rpS6 in these cells effectively downregulated G1 phase-related proteins, including RB, cyclin D1, and CDK6, induced cell cycle arrest, and inhibited proliferation, suggesting that aberrant modulation of rpS6 phosphorylation contributed to the acquired resistance. Interestingly, RSK inhibitor had little effect on rpS6 phosphorylation and cell proliferation in resistant cells, whereas P70S6K inhibitor showed stronger inhibitory effects on rpS6 phosphorylation and cell proliferation in resistant cells than in parental cells. Thus regulation of rpS6 phosphorylation, which is predominantly mediated by BRAF/MEK/ERK/RSK signaling in parental cells, was switched to mTOR/ P70S6K signaling in resistant cells. Furthermore, mTOR inhibitors alone overcame acquired resistance and rescued the sensitivity of the resistant cells when combined with BRAF/MEK inhibitors. -

Human Melanoma Cells Resistant to MAPK Inhibitors Can Be Effectively Targeted by Inhibition of the P90 Ribosomal S6 Kinase
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 22), pp: 35761-35775 Research Paper Human melanoma cells resistant to MAPK inhibitors can be effectively targeted by inhibition of the p90 ribosomal S6 kinase Corinna Kosnopfel1, Tobias Sinnberg1, Birgit Sauer1, Heike Niessner1, Anja Schmitt2, Elena Makino1, Andrea Forschner1, Stephan Hailfinger2, Claus Garbe1, Birgit Schittek1 1Division of Dermatooncology, Department of Dermatology, University of Tübingen, Tübingen, Germany 2Interfaculty Institute of Biochemistry, University of Tübingen, Tübingen, Germany Correspondence to: Birgit Schittek, email: [email protected] Keywords: melanoma, MAPK inhibition, therapy resistance, p90 ribosomal S6 kinase, YB-1 Received: January 18, 2017 Accepted: March 06, 2017 Published: March 15, 2017 Copyright: Kosnopfel et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC-BY), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT The clinical availability of small molecule inhibitors specifically targeting mutated BRAF marked a significant breakthrough in melanoma therapy. Despite a dramatic anti-tumour activity and improved patient survival, rapidly emerging resistance, however, greatly limits the clinical benefit. The majority of the already described resistance mechanisms involve a reactivation of the MAPK signalling pathway. The p90 ribosomal S6 kinase (RSK), a downstream effector of the MAPK signalling cascade, has been reported to enhance survival of melanoma cells in response to chemotherapy. Here, we can show that RSK activity is significantly increased in human melanoma cells with acquired resistance to the BRAFV600E/K inhibitor vemurafenib. Interestingly, inhibition of RSK signalling markedly impairs the viability of vemurafenib resistant melanoma cells and is effective both in two-dimensional and in three-dimensional culture systems, especially in a chronic, long-term application. -

Supplementary Information Material and Methods
MCT-11-0474 BKM120: a potent and specific pan-PI3K inhibitor Supplementary Information Material and methods Chemicals The EGFR inhibitor NVP-AEE788 (Novartis), the Jak inhibitor I (Merck Calbiochem, #420099) and anisomycin (Alomone labs, # A-520) were prepared as 50 mM stock solutions in 100% DMSO. Doxorubicin (Adriablastin, Pfizer), EGF (Sigma Ref: E9644), PDGF (Sigma, Ref: P4306) and IL-4 (Sigma, Ref: I-4269) stock solutions were prepared as recommended by the manufacturer. For in vivo administration: Temodal (20 mg Temozolomide capsules, Essex Chemie AG, Luzern) was dissolved in 4 mL KZI/glucose (20/80, vol/vol); Taxotere was bought as 40 mg/mL solution (Sanofi Aventis, France), and prepared in KZI/glucose. Antibodies The primary antibodies used were as follows: anti-S473P-Akt (#9271), anti-T308P-Akt (#9276,), anti-S9P-GSK3β (#9336), anti-T389P-p70S6K (#9205), anti-YP/TP-Erk1/2 (#9101), anti-YP/TP-p38 (#9215), anti-YP/TP-JNK1/2 (#9101), anti-Y751P-PDGFR (#3161), anti- p21Cip1/Waf1 (#2946), anti-p27Kip1 (#2552) and anti-Ser15-p53 (#9284) antibodies were from Cell Signaling Technologies; anti-Akt (#05-591), anti-T32P-FKHRL1 (#06-952) and anti- PDGFR (#06-495) antibodies were from Upstate; anti-IGF-1R (#SC-713) and anti-EGFR (#SC-03) antibodies were from Santa Cruz; anti-GSK3α/β (#44610), anti-Y641P-Stat6 (#611566), anti-S1981P-ATM (#200-301), anti-T2609 DNA-PKcs (#GTX24194) and anti- 1 MCT-11-0474 BKM120: a potent and specific pan-PI3K inhibitor Y1316P-IGF-1R were from Bio-Source International, Becton-Dickinson, Rockland, GenTex and internal production, respectively. The 4G10 antibody was from Millipore (#05-321MG). -

The Role of the S6K2 Splice Isoform in Mtor/S6K Signalling and Cellular Functions
The role of the S6K2 splice isoform in mTOR/S6K signalling and cellular functions Olena Myronova A thesis submitted to the University College London in fulfilment with the requirements for the degree of Doctor of Philosophy London, November 2015 Research Department of Structural and Molecular Biology Division of Biosciences University College London Gower Street London, WC1E 6BT United Kingdom Ludwig Institute for Cancer Research 666 Third Avenue, 28th floor New York, N.Y. 10017 USA The role of the S6K2 splice isoform in mTOR/S6K signalling and cellular functions 1 Declaration I, Olena Myronova, declare that all the work presented in this thesis is the result of my own work. The work presented here does not constitute part of any other thesis. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. The work here in was carried out while I was a graduate research student at University College London, Research Department of Structural and Molecular Biology under the supervision of Professor Ivan Gout. Olena Myronova The role of the S6K2 splice isoform in mTOR/S6K signalling and cellular functions 2 Abstract Ribosomal S6 kinase (S6K) is a member of the AGC family of serine/threonine protein kinases and plays a key role in diverse cellular processes, including cell growth, survival and metabolism. Activation of S6K by growth factors, amino acids, energy levels and hypoxia is mediated by the mTOR and PI3K signalling pathways. Dysregulation of S6K activity has been implicated in a number of human pathologies, including cancer, diabetes, obesity and ageing. -
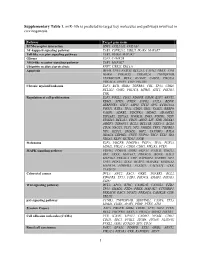
1 Supplementary Table 1. Mir-10B Is Predicted to Target Key Molecules
Supplementary Table 1. miR-10b is predicted to target key molecules and pathways involved in carcinogenesis. Pathway Target gene name ECM-receptor interaction SDC1, COL24A1, COL4A4 NF-kappa B signaling pathway TAB1, CSNK2A2, UBE2I, IRAK4, MAP3K7 Toll-like receptor signaling pathway TAB1, IRAK4, MAP3K7 Glioma E2F3, CAMK2B NOD-like receptor signaling pathway TAB1, MAP3K7 Ubiquitin mediated proteolysis RNF7, UBE2I, ERCC8 Apoptosis DFFB, TP53, FASLG, BCL2L1, CAPN2, PRKX, ATM, IRAK4, PRKACG, PRKAR2A, TNFRSF10B, TNFRSF10D, BCL2, IL1RAP, CASP8, PIK3CA, PRKACA, APAF1, CHP, PIK3R3 Chronic myeloid leukemia E2F3, BCR, GRB2, TGFBR1, CBL, TP53, CDK6, BCL2L1, GAB2, PIK3CA, MDM2, SHC1, PIK3R3, CRK Regulation of cell proliferation E2F3, FOSL2, CDX2, PDGFB, OSMR, E2F7, ARNT2, RBM5, STRN, PTEN, S1PR2, CUL3, BDNF, SERPINE1, SHC1, ASPH, ITCH, SPN, CCDC88A, FOXJ1, RXRA, TP53, CDK6, IRS1, VASH2, RBBP9, VASH1, ADRB2, PDGFRA, MDM2, ADAMTS1, EIF2AK2, EIF5A2, ICOSLG, ING5, FGFR3, NDN, ST8SIA1, BCL2L1, CDH5, ARNT, LIF, VDR, HOXA3, AGGF1, TSPAN31, BCL2, BCL11B, NKX3-1, BCL6, CD28, NACC1, FLT1, NF2, JARID2, TBX5, TGFBR1, NF1, KLF11, SMAD2, IGF2, TAX1BP3, BTLA, HDAC4, LEPRE1, CNTF, NUP62, TSC1, ETS1, ID4, NR5A2, KLF4, KCTD11, NFIB Melanoma E2F3, PDGFB, PDGFRA, FGF11, TP53, FGF23, MDM2, PIK3CA, CDK6, CDH1, PIK3R3, PTEN MAPK signaling pathway FGFR3, PDGFB, GRB2, FGF11, FASLG, GNG12, SRF, PRKX, MAP3K7, PRKACG, BDNF, RAC3, MAP3K2, PRKACA, CHP, RAPGEF2, TGFBR1, NF1, TP53, FGF23, STK4, DUSP5, MAP4K4, RPS6KA2, MAPK14, PDGFRA, PLA2G3, CACNA1C, CRK, PLA2G2F Colorectal cancer -

In Vitro Pharmacological Profiling of R406 Identifies Molecular Targets
ORIGINAL ARTICLE In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib Michael G. Rolf, Jon O. Curwen, Margaret Veldman-Jones, Cath Eberlein, Jianyan Wang, Alex Harmer, Caroline J. Hellawell & Martin Braddock AstraZeneca R&D Alderley Park, Macclesfield, Cheshire SK10 4TG, United Kingdom Keywords Abstract Blood pressure elevation, fostamatinib, in vitro pharmacological profiling, R406, SYK Off-target pharmacology may contribute to both adverse and beneficial effects of a new drug. In vitro pharmacological profiling is often applied early in drug Correspondence discovery; there are fewer reports addressing the relevance of broad profiles to Michael G. Rolf, AstraZeneca R&D Molndal,€ clinical adverse effects. Here, we have characterized the pharmacological profile Pepparedsleden 1, 431 83 Mo¨ lndal, Sweden. of the active metabolite of fostamatinib, R406, linking an understanding of drug Tel: +46 31 776 60 40; Fax: +46 31 776 37 selectivity to the increase in blood pressure observed in clinical studies. R406 60; E-mail: [email protected] was profiled in a broad range of in vitro assays to generate a comprehensive Funding Information pharmacological profile and key targets were further investigated using func- No funding information provided. tional and cellular assay systems. A combination of traditional literature searches and text-mining approaches established potential mechanistic links Received: 9 April 2015; Accepted: 14 July between the profile of R406 and clinical side effects. R406 was selective outside 2015 the kinase domain, with only antagonist activity at the adenosine A3 receptor in the range relevant to clinical effects. R406 was less selective in the kinase Pharma Res Per, 3(5), 2015, e00175, doi: 10.1002/prp2.175 domain, having activity at many protein kinases at therapeutically relevant con- centrations when tested in multiple in vitro systems. -

Resistance to Drugs and Cell Death in Cancer Stem Cells (Cscs)
Journal of Translational Science Review Article ISSN: 2059-268X Resistance to drugs and cell death in cancer stem cells (CSCs) Ahmad R Safa* Department of Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, USA Abstract Human cancers emerge from cancer stem cells (CSCs), which are resistant to cancer chemotherapeutic agents, radiation, and cell death. Moreover, autophagy provides the cytoprotective effect which contributes to drug resistance in these cells. Furthermore, much evidence shows that CSCs cause tumor initiation, progression, metastasis, and cancer recurrence. Various signaling pathways including the phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR), maternal embryonic leucine zipper kinase (MELK), NOTCH1, and Wnt/β-catenin as well as the CSC markers maintain CSC properties. Several mechanisms including overexpression of ABC multidrug resistance transporters, a deficiency in mitochondrial-mediated apoptosis, upregulation of c-FLIP, overexpression of anti- apoptotic Bcl-2 family members and inhibitors of apoptosis proteins (IAPs), and PI3K/AKT signaling contribute to enhancing resistance to chemotherapeutic drugs and cell death induction in CSCs in various cancers. Studying such pathways may help provide detailed understanding of CSC mechanisms of resistance to chemotherapeutic agents and apoptosis and may lead to the development of effective therapeutics to eradicate CSCs. Abbreviations: Apaf-1: Apoptotic Proteinase-Activating Factor-1; Tumor recurrence is the major cause of death in patients with AIC: Apoptosis Inhibitory Complex; BH3: Bcl-2 Homology Domain-3; incurable malignancies and is attributed to treatment-resistant CSCs CSCs: Cancer Stem Cells; c-FLIP: cellular FLICE (FADD-like IL- within the primary tumor (Figure 2). CSCs are a rare cell population 1β-converting enzyme)-Inhibitory Protein; DISC: Death-Inducing within a tumor, which express differing molecular markers in various Signaling Complex; DNA-PK: DNA-Dependent Protein Kinase; FADD: types of cancers [1,2,4]. -

Extracellular Receptor Kinase and Camp Response Element Binding Protein Activation in the Neonatal Rat Heart After Perinatal Cocaine Exposure
0031-3998/04/5606-0947 PEDIATRIC RESEARCH Vol. 56, No. 6, 2004 Copyright © 2004 International Pediatric Research Foundation, Inc. Printed in U.S.A. Extracellular Receptor Kinase and cAMP Response Element Binding Protein Activation in the Neonatal Rat Heart after Perinatal Cocaine Exposure LENA S. SUN AND AARON QUAMINA Department of Anesthesiology [L.S.S., A.Q.] and Pediatrics [L.S.S.], College of Physicians & Surgeons, Columbia University, New York, NY 10032 ABSTRACT Prenatal exposure to cocaine has been shown to induce an and phospho-RSK. We assessed the interaction of RSK with increase in the myocardial expression and activation of the CREB or CREB-binding protein by performing co-immunopre- cAMP response binding protein (CREB), a transcriptional factor cipitation experiments. We found that perinatal cocaine exposure that has been shown to regulate gene expression. Several differ- increased both phospho-ERK and phospho-RSK expression, in- ent kinases, including protein kinase A, calcium calmodulin dicative of an increased activity of these two enzymes. Further- kinase II, and mitogen-activated protein kinase can induce phos- more, we demonstrated that phospho-RSK was immunoprecipi- phorylation of CREB at serine 133, a necessary step for CREB tated with CREB in all neonatal cardiac nuclei and that the activation. We examined whether the mitogen-activated protein greatest interaction was found in day 7 hearts after perinatal kinase–extracellular receptor kinase (ERK) pathway may be cocaine exposure. Our results thus illustrate that the ERK-RSK involved in mediating the serine 133 CREB phosphorylation in pathway was active in the postnatal rat heart at 1 and7dofage cardiac nuclei after perinatal cocaine exposure. -

Phospho-RPS6KA1(T359) Antibody Peptide Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap3497a
9765 Clairemont Mesa Blvd, Suite C San Diego, CA 92124 Tel: 858.875.1900 Fax: 858.622.0609 Phospho-RPS6KA1(T359) Antibody Peptide Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP3497a Specification Phospho-RPS6KA1(T359) Antibody - Product Information Application WB, DB,E Primary Accession Q15418 Other Accession P10666, P10665, P18652 Reactivity Human Predicted Chicken, Xenopus Host Rabbit Clonality Polyclonal Isotype Rabbit Ig Calculated MW 82723 Phospho-RPS6KA1(T359) Antibody - Additional Information Gene ID 6195 Other Names Western blot analysis of extracts from Hela Ribosomal protein S6 kinase alpha-1, cells,untreated or treated with S6K-alpha-1, 90 kDa ribosomal protein S6 TPA,200nM,using phospho RPS6KA1-T359 (left) kinase 1, p90-RSK 1, p90RSK1, p90S6K, MAP or RPS6KA1 antibody(right) kinase-activated protein kinase 1a, MAPK-activated protein kinase 1a, MAPKAP kinase 1a, MAPKAPK-1a, Ribosomal S6 kinase 1, RSK-1, RPS6KA1, MAPKAPK1A, RSK1 Target/Specificity This RPS6KA1 Antibody is generated from rabbits immunized with a KLH conjugated synthetic phosphopeptide corresponding to amino acid residues surrounding T359 of human RPS6KA1. Dilution WB~~1:2000 DB~~1:500 Format Purified polyclonal antibody supplied in PBS with 0.09% (W/V) sodium azide. This antibody is purified through a protein A column, followed by peptide affinity purification. Dot blot analysis of anti-RPS6KA1-pT359 Pab (RB13385) on nitrocellulose membrane. 50ng Storage of Phospho-peptide or Non Phospho-peptide per Maintain refrigerated at 2-8°C for up to 6 dot were adsorbed. Antibody working months. For long term storage store at -20°C concentrations are 0.5ug per ml. in small aliquots to prevent freeze-thaw cycles. -

Autophagy Processes Are Dependent on EGF Receptor Signaling
www.oncotarget.com Oncotarget, 2018, Vol. 9, (No. 54), pp: 30289-30303 Research Paper Autophagy processes are dependent on EGF receptor signaling Vincenzo De Iuliis1, Antonio Marino1, Marika Caruso1, Sabrina Capodifoglio1, Vincenzo Flati2, Anna Marynuk3, Valeria Marricareda3, Sebastiano Ursi1, Paola Lanuti4, Claudio Talora5, Pio Conti6, Stefano Martinotti1,7 and Elena Toniato7 1Unit of Predictive Medicine, SS Annunziata University Hospital of Chieti, Chieti, Italy 2Department of Biotechnological and Applied Clinical Sciences, University of L’Aquila, L’Aquila, Italy 3Odessa National Medical University, Odesa, Odessa Oblsat, Ucraina 4Department of Medicine and Aging Sciences, University G. d’Annunzio of Chieti, Chieti, Italy 5Department of Molecular Medicine, University of Rome “La Sapienza”, Rome, Italy 6Postgraduate Medical School, University of Chieti, Chieti, Italy 7Department of Medical, Oral and Biotechnological Sciences, University G. d’Annunzio of Chieti, Chieti, Italy Correspondence to: Elena Toniato, email: [email protected] Keywords: apoptosis; autophagy; Beclin 1; EGF receptor; MAPK pathway Received: May 03, 2018 Accepted: June 13, 2018 Published: July 13, 2018 Copyright: De Iuliis et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Autophagy is a not well-understood conserved mechanism activated during nutritional deprivation in order to maintain cellular homeostasis. In the present study, we investigated the correlations between autophagy, apoptosis and the MAPK pathways in melanoma cell lines. We demonstrated that during starvation the EGF receptor mediated signaling activates many proteins involved in the MAPK pathway. -

DAPK) Through a Lysosome-Dependent Degradation Pathway Yao Lin1, Paul Henderson1,2, Susanne Pettersson1, Jack Satsangi1, Ted Hupp1 and Craig Stevens1
Tuberous sclerosis-2 (TSC2) regulates the stability of death-associated protein kinase-1 (DAPK) through a lysosome-dependent degradation pathway Yao Lin1, Paul Henderson1,2, Susanne Pettersson1, Jack Satsangi1, Ted Hupp1 and Craig Stevens1 1 University of Edinburgh, Institute of Genetics and Molecular Medicine, UK 2 Department of Child Life and Health, University of Edinburgh, UK Keywords We previously identified a novel interaction between tuberous sclerosis-2 DAPK; degradation; lysosome; mTORC1; (TSC2) and death-associated protein kinase-1 (DAPK), the consequence TSC2 being that DAPK catalyses the inactivating phosphorylation of TSC2 to stimulate mammalian target of rapamycin complex 1 (mTORC1) activity. Correspondence C. Stevens, University of Edinburgh, We now report that TSC2 binding to DAPK promotes the degradation of Institute of Genetics and Molecular DAPK. We show that DAPK protein levels, but not gene expression, Medicine, Edinburgh, EH4 2XR, UK inversely correlate with TSC2 expression. Furthermore, altering mTORC1 Fax: +44 131 651 1085 activity does not affect DAPK levels, excluding indirect effects of TSC2 on Tel: +44 131 651 1025 DAPK protein levels through changes in mTORC1 translational control. E-mail: [email protected] We provide evidence that the C-terminus regulates TSC2 stability and is required for TSC2 to reduce DAPK protein levels. Importantly, using a (Received 28 July 2010, revised 7 October 2010, accepted 11 November 2010) GTPase-activating protein–dead missense mutation of TSC2, we demon- strate that the effect of TSC2 on DAPK is independent of GTPase-activat- doi:10.1111/j.1742-4658.2010.07959.x ing protein activity. TSC2 binds to the death domain of DAPK and we show that this interaction is required for TSC2 to reduce DAPK protein levels and half-life.