14-3-3 Proteins Inactivate DAPK2 by Promoting Its Dimerization And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Novel Functions of Death-Associated Protein Kinases Through Mitogen-Activated Protein Kinase-Related Signals
International Journal of Molecular Sciences Article Novel Functions of Death-Associated Protein Kinases through Mitogen-Activated Protein Kinase-Related Signals Mohamed Elbadawy 1,2,† , Tatsuya Usui 1,*,†, Hideyuki Yamawaki 3 and Kazuaki Sasaki 1 1 Laboratory of Veterinary Pharmacology, Department of Veterinary Medicine, Faculty of Agriculture, Tokyo University of Agriculture and Technology, 3-5-8 Saiwai-cho, Fuchu, Tokyo 183-8509, Japan; [email protected] (M.E.); [email protected] (K.S.) 2 Department of Pharmacology, Faculty of Veterinary Medicine, Benha University, Moshtohor, Elqaliobiya, Toukh 13736, Egypt 3 Laboratory of Veterinary Pharmacology, School of Veterinary Medicine, Kitasato University, Towada, Aomori 034-8628, Japan; [email protected] * Correspondence: [email protected]; Tel./Fax: +81-42-367-5769 † These authors contributed equally to this work. Received: 13 September 2018; Accepted: 1 October 2018; Published: 4 October 2018 Abstract: Death associated protein kinase (DAPK) is a calcium/calmodulin-regulated serine/threonine kinase; its main function is to regulate cell death. DAPK family proteins consist of DAPK1, DAPK2, DAPK3, DAPK-related apoptosis-inducing protein kinases (DRAK)-1 and DRAK-2. In this review, we discuss the roles and regulatory mechanisms of DAPK family members and their relevance to diseases. Furthermore, a special focus is given to several reports describing cross-talks between DAPKs and mitogen-activated protein kinases (MAPK) family members in various pathologies. We also discuss small molecule inhibitors of DAPKs and their potential as therapeutic targets against human diseases. Keywords: MAPK; DAPK; ERK; p38; JNK 1. Introduction: DAPKs, MAPKs Death-associated protein kinase (DAPK) family proteins are closely related, Ca2+/calmodulin (CaM)-regulated serine/threonine kinases, whose members not only possess significant homology in their catalytic domains but also share cell death-associated functions [1,2]. -

Anti-DAPK2 Antibody (ARG54388)
Product datasheet [email protected] ARG54388 Package: 50 μg anti-DAPK2 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes DAPK2 Tested Reactivity Hu, Ms, Rat Tested Application IHC, WB Specificity This antibody recognizes human, mouse, and rat DAPK2 (approx. 42kDa) and does not cross-react with DAPK. Host Rabbit Clonality Polyclonal Isotype IgG Target Name DAPK2 Antigen Species Human Immunogen Peptide corresponding to aa 356-370 of human DAPK2 (accession no. BAA88063). This sequence is identical to that of mouse. Conjugation Un-conjugated Alternate Names DRP1; DAP-kinase-related protein 1; DRP-1; DAP kinase 2; EC 2.7.11.1; Death-associated protein kinase 2 Application Instructions Application table Application Dilution IHC Assay-dependent WB Assay-dependent Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control A431, Mouse spleen and Rat kidney Calculated Mw 43 kDa Properties Form Liquid Purification Immunoaffinity chroma-tography Buffer PBS (pH 7.4) and 0.02% Sodium azide Preservative 0.02% Sodium azide Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C or below. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed www.arigobio.com 1/3 before use. Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Database links GeneID: 13143 Mouse GeneID: 23604 Human Swiss-port # Q8VDF3 Mouse Swiss-port # Q9UIK4 Human Gene Symbol DAPK2 Gene Full Name death-associated protein kinase 2 Background Certain serine/threonine protein kinases, such as RIP and DAP kinase, are mediators of apoptosis. -
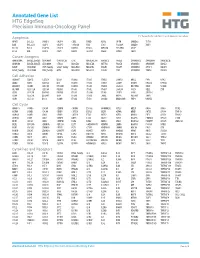
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain -

Gene Section Review
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL INIST-CNRS Gene Section Review DAPK2 (death-associated protein kinase 2) Mafalda Pinto, Valdemar Máximo IPATIMUP Institute of Molecular Pathology and Immunology of the University of Porto, (MP, VM); I3S Institute for Innovation and Health Research, University of Porto (MP, VM); Department of Pathology and Oncology, Medical Faculty of the University of Porto, Porto, Portugal (VM) [email protected]; [email protected] Published in Atlas Database: April 2016 Online updated version : http://AtlasGeneticsOncology.org/Genes/DAPK2ID40263ch15q22.html Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/66944/04-2016-DAPK2ID40263ch15q22.pdf DOI: 10.4267/2042/66944 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2016 Atlas of Genetics and Cytogenetics in Oncology and Haematology Location (base pair): Chromosome 15:63,907,036- Abstract 64,072,033 reverse strand. GRCh38:CM000677.2 (Ensembl.org) Short communication on DAPK2, with data on DNA and on the protein encoded. DNA/RNA Keywords DAPK2; DRP1; DRP-1; calcium/calmodulin; DAPK2 is a gene that codes for a protein that serine/threonine; kinase; apoptosis belongs to the serine/threonine protein kinase family. Identity Transcription Other names: DRP1, DRP-1 DPAK2 has 13 transcripts (3 coding), 75 orthologues and 11 paralogues (MYLK4, MYLK3, STK17A, HGNC (Hugo): DAPK2 DAPK3, DAPK1, OBSCN, SPEG, TTN, MYLK, Location: 15q22.31 STK17B, MYKL2) (ENSG00000035664). Blue highlighting indicates alternating exons; Red highlighting indicates amino acids encoded across a splice junction. Atlas Genet Cytogenet Oncol Haematol. 2016; 20(12) 587 DAPK2 (death-associated protein kinase 2) Pinto M, Máximo V Schematic diagram of DAPK2 protein structure. -

BCL6 Confers KRAS-Mutant Non–Small-Cell Lung Cancer Resistance to BET Inhibitors
BCL6 confers KRAS-mutant non–small-cell lung cancer resistance to BET inhibitors Jiawei Guo, … , Mingyao Liu, Xiufeng Pang J Clin Invest. 2021;131(1):e133090. https://doi.org/10.1172/JCI133090. Research Article Oncology Graphical abstract Find the latest version: https://jci.me/133090/pdf The Journal of Clinical Investigation RESEARCH ARTICLE BCL6 confers KRAS-mutant non–small-cell lung cancer resistance to BET inhibitors Jiawei Guo,1,2 Yanan Liu,1 Jing Lv,1 Bin Zou,3 Zhi Chen,1,4 Kun Li,1 Juanjuan Feng,1 Zhenyu Cai,5 Lai Wei,3 Mingyao Liu,1 and Xiufeng Pang1 1Shanghai Key Laboratory of Regulatory Biology, Institute of Biomedical Sciences and School of Life Sciences, East China Normal University, Shanghai, China. 2Department of Thoracic Surgery, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University and Collaborative Innovation Center for Biotherapy, Chengdu, China. 3State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China. 4Medical Research Institute, Wuhan University, Wuhan, China. 5Tongji University Cancer Center, Shanghai Tenth People’s Hospital, School of Medicine, Tongji University, Shanghai, China. The bromodomain and extra-terminal domain (BET) proteins are promising therapeutic targets to treat refractory solid tumors; however, inherent resistance remains a major challenge in the clinic. Recently, the emerging role of the oncoprotein B cell lymphoma 6 (BCL6) in tumorigenesis and stress response has been unveiled. Here, we demonstrate that BCL6 was upregulated upon BET inhibition in KRAS-mutant cancers, including non–small-cell lung cancer (NSCLC). We further found that BRD3, not BRD2 or BRD4, directly interacted with BCL6 and maintained the negative autoregulatory circuit of BCL6. -

Polymorphisms Within Autophagy-Related Genes Influence the Risk of Developing Colorectal Cancer: a Meta-Analysis of Four Large Cohorts
cancers Article Polymorphisms within Autophagy-Related Genes Influence the Risk of Developing Colorectal Cancer: A Meta-Analysis of Four Large Cohorts Juan Sainz 1,2,3,4,* , Francisco José García-Verdejo 5, Manuel Martínez-Bueno 6 , Abhishek Kumar 7,8,9 , José Manuel Sánchez-Maldonado 1,2,3, Anna Díez-Villanueva 10 , Ludmila Vodiˇcková 11,12,13, Veronika Vymetálková 11,12,13 , Vicente Martin Sánchez 14,15 , Miguel Inacio Da Silva Filho 7, Belém Sampaio-Marques 16,17, Stefanie Brezina 18, Katja Butterbach 19, Rob ter Horst 20, Michael Hoffmeister 21, Paula Ludovico 16,17 , Manuel Jurado 1,2,3, Yang Li 20,22 , Pedro Sánchez-Rovira 5, Mihai G. Netea 20,23, Andrea Gsur 18 , Pavel Vodiˇcka 11,12,13,Víctor Moreno 10, Kari Hemminki 7,24,25, Hermann Brenner 19,26,27 , Jenny Chang-Claude 28,29 and Asta Försti 7,30,31,* 1 Genomic Oncology Area, GENYO, Centre for Genomics and Oncological Research Pfizer/University of Granada/Andalusian Regional Government, 18016 Granada, Spain; [email protected] (J.M.S.-M.); [email protected] (M.J.) 2 Hematology Department, Virgen de las Nieves University Hospital, 18012 Granada, Spain 3 Instituto de Investigación Biosanataria IBs.Granada, 18012 Granada, Spain 4 Department of Medicine, University of Granada, 18016 Granada, Spain Citation: Sainz, J.; García-Verdejo, 5 Department of Medical Oncology, Complejo Hospitalario de Jaén, 23007 Jaén, Spain; F.J.; Martínez-Bueno, M.; Kumar, A.; [email protected] (F.J.G.-V.); [email protected] (P.S.-R.) Sánchez-Maldonado, J.M.; 6 Area of Genomic Medicine, GENYO, Centre for Genomics and Oncological Research Pfizer/University of Díez-Villanueva, A.; Vodiˇcková,L.; Granada/Andalusian Regional Government, 18016 Granada, Spain; [email protected] Vymetálková, V.; Martin Sánchez, V.; 7 Division of Molecular Genetic Epidemiology, German Cancer Research Center (DKFZ), Da Silva Filho, M.I.; et al. -

Development and Validation of a Robust Ferroptosis-Related Prognostic Signature in Lung Adenocarcinoma
fcell-09-616271 June 23, 2021 Time: 12:21 # 1 ORIGINAL RESEARCH published: 24 June 2021 doi: 10.3389/fcell.2021.616271 Development and Validation of a Robust Ferroptosis-Related Prognostic Signature in Lung Adenocarcinoma Anran Zhang1†, Jinpo Yang2†, Chao Ma3,4*, Feng Li3,5 and Huan Luo3 1 Department of Oncology, Henan Provincial People’s Hospital, Zhengzhou University People’s Hospital, Henan University People’s Hospital, Zhengzhou, China, 2 Department of Medical Oncology, The Affiliated Cancer Hospital of Zhengzhou University, Henan Cancer Hospital, Zhengzhou, China, 3 Charité – Universitätsmedizin Berlin, Freie Universität Berlin, Humboldt-Universität zu Berlin, Berlin Institute of Health, Berlin, Germany, 4 Berlin Institute of Health Center for Regenerative Edited by: Therapies and Berlin-Brandenburg Center for Regenerative Therapies (BCRT), Charité – Universitätsmedizin Berlin, Berlin, Jawed Akhtar Siddiqui, Germany, 5 Department of Surgery, Competence Center of Thoracic Surgery, Charité University Hospital Berlin, Berlin, University of Nebraska Medical Germany Center, United States Reviewed by: Tao Jiang, Background: Lung adenocarcinoma (LUAD) is the most common subtype of non- University of Illinois small cell lung cancer. Ferroptosis is a newly recognized process of cell death, which is at Urbana-Champaign, United States Concetta Saponaro, different from other forms of cell death in morphology, biochemistry, and genetics, and Istituto Tumori Bari Giovanni Paolo II, has played a vital role in cancer biology. This study aimed to identify a ferroptosis-related Istituto Nazionale dei Tumori (IRCCS), gene signature associated with LUAD prognosis. Italy *Correspondence: Methods: Dataset TCGA-LUAD which came from the TCGA portal was taken as Chao Ma the training cohort. GSE72094 and GSE68465 from the GEO database were treated [email protected] orcid.org/0000-0003-1444-4668 as validation cohorts. -

Oligonucleotide Microarray Analysis of Distinct Gene Expression Patterns in Colorectal Cancer Tissues Harboring BRAF and K-Ras Mutations
Carcinogenesis vol.27 no.3 pp.392–404, 2006 doi:10.1093/carcin/bgi237 Advance Access publication October 11, 2005 Oligonucleotide microarray analysis of distinct gene expression patterns in colorectal cancer tissues harboring BRAF and K-ras mutations Il-Jin Kim1,y, Hio Chung Kang1,2,y, Sang-Geun Jang1, Introduction Kun Kim1, Sun-A Ahn1, Hyun-Ju Yoon1, Sang Nam Yoon3 and Jae-Gahb Park1,2,3,Ã Colorectal cancer (CRC) is an important human cancer, and incidence rates of this cancer are increasing in Asian countries 1 Korean Hereditary Tumor Registry, Cancer Research Institute and such as Korea (1). CRC development is a multi-step process Downloaded from https://academic.oup.com/carcin/article/27/3/392/2476120 by guest on 27 September 2021 Cancer Research Center, Seoul National University, Seoul, Korea, (2) involving microsatellite instability (MSI), mutations in 2Research Institute and Hospital, National Cancer Center, 809 Madu-dong, Ilsan-gu, Goyang, Gyeonggi 411-764, Korea mismatch repair (MMR) of genes such as MLH1 and MSH2, and 3Department of Surgery, Seoul National University College of and mutations in APC, SMAD4,K-ras, TP53 and b-catenin Medicine, Seoul, Korea (2–9). Somatic BRAF mutations have been reported in ÃTo whom correspondence should be addressed. 5.1–18% of CRCs (10–12). BRAF is one of three serine/ E-mail: [email protected] threonine kinases (ARAF, BRAF, CRAF/RAF1) that act within Various types of human cancers harbor BRAF somatic the RAS–RAF–MEK–ERK–MAPK signaling pathway (13). mutations, leading researchers to seek molecular targets BRAF mutations have also been reported in 80% of primary for BRAF inhibitors. -

BRAFV600E-Mutant Cancers Display a Variety of Networks by SWIM Analysis: Prediction of Vemurafenib Clinical Response
Endocrine (2019) 64:406–413 https://doi.org/10.1007/s12020-019-01890-4 ORIGINAL ARTICLE BRAFV600E-mutant cancers display a variety of networks by SWIM analysis: prediction of vemurafenib clinical response 1 2,3 2,3 1 1 Rosa Falcone ● Federica Conte ● Giulia Fiscon ● Valeria Pecce ● Marialuisa Sponziello ● 1 4 1 2 1 Cosimo Durante ● Lorenzo Farina ● Sebastiano Filetti ● Paola Paci ● Antonella Verrienti Received: 30 January 2019 / Accepted: 1 March 2019 / Published online: 8 March 2019 © Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Purpose Several studies have shown that different tumour types sharing a driver gene mutation do not respond uniformly to the same targeted agent. Our aim was to use an unbiased network-based approach to investigate this fundamental issue using BRAFV600E mutant tumours and the BRAF inhibitor vemurafenib. Methods We applied SWIM, a software able to identify putative regulatory (switch) genes involved in drastic changes to the cell phenotype, to gene expression profiles of different BRAFV600E mutant cancers and their normal counterparts in order to identify the switch genes that could potentially explain the heterogeneity of these tumours’ responses to vemurafenib. fi 1234567890();,: 1234567890();,: Results We identi ed lung adenocarcinoma as the tumour with the highest number of switch genes (298) compared to its normal counterpart. By looking for switch genes encoding for kinases with homology sequences similar to known vemurafenib targets, we found that thyroid cancer and lung adenocarcinoma have a similar number of putative targetable switch gene kinases (5 and 6, respectively) whereas colorectal cancer has just one. Conclusions We are persuaded that our network analysis may aid in the comprehension of molecular mechanisms under- lying the different responses to vemurafenib in BRAFV600E mutant tumours.