Umpolung Reactions at the Α-Carbon Position of Carbonyl Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UMPOLUNG in REACTIONS CATALYZED by THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung En Reacciones Catalizadas Por Enzimas Dependientes De Pirofosfato De Tiamina
Ciencia, Ambiente y Clima, Vol. 2, No. 2, julio-diciembre, 2019 • ISSN (impreso): 2636-2317 • ISSN (en línea): 2636-2333 DOI: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 UMPOLUNG IN REACTIONS CATALYZED BY THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung en reacciones catalizadas por enzimas dependientes de pirofosfato de tiamina Carlos José Boluda Emily Soto Instituto Tecnológico de Santo Domingo (INTEC), Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales, Av. de Los Área de Ciencias Básicas y Ambientales Próceres 49, Santo Domingo, República Dominicana Correo-e: [email protected] *Corresponding author: Carlos J. Boluda Darah de la Cruz Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Carolina Juncá Área de Ciencias Básicas y Ambientales Instituto Tecnológico de Santo Domingo (INTEC), Correo-e: [email protected] Área de Ciencias Básicas y Ambientales Anny Peña Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales Correo-e: [email protected] Recibido: 25/9/2019 • Aprobado: 19/10/2019 Cómo citar: Boluda, C. J., Juncá, C., Soto, E., de la Cruz, D., & Peña, A. (2019). Umpolung in reactions catalyzed by thiamine pyrophos- phate dependent enzymes. Ciencia, Ambiente Y Clima, 2(2), 27-42. Doi: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 Abstract Resumen The temporal exchange of the electrophilic/nucleophilic El intercambio temporal del carácter electrofílico/nucleofí- character of an atom by chemical manipulation is known lico de un átomo mediante manipulación química, es cono- in organic chemistry as umpolung. This inversion of polarity cido con el vocablo alemán de umpolung. -
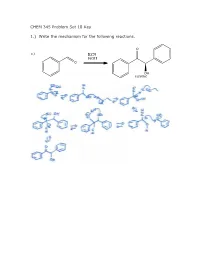
CHEM 345 Problem Set 18 Key 1.) Write the Mechanism for The
CHEM 345 Problem Set 18 Key 1.) Write the mechanism for the following reactions. O a.) KCN EtOH O OH racemic 1.) Write the mechanism for the following reactions. b.) KCN AcOH O NC OH racemic O c.) N S R O NEt3 OH racemic 2.) What is the structure of AcOH?Why does changing the solvent from EtOH to AcOH make such a big difference? O OH AcOH acetic acid The pKa of acetic acid is approximately 5. The pKa of ethanol is approximately 15. When you take a proton off of ethanol, you generate ethoxide which is about 1010 times stronger of a base than acetate. 3.) Give two instances when you need to use the thiazolium salt and triethylamine rather than KCN and EtOH. If the aldehydes contain an enolizable proton then you cannot use KCN/EtOH, instead you must use the thiazolium. Also, if the electrophile is a Michael acceptor to give a 1,4 dicarbonyl, then the thiazolium catalyst should be used. 4.) Break the following compound down as far as you can using Aldol, Michael, and Claisen reactions. Above each retrosynthetic arrow, write the name of the reaction. O HO O HO Aldol O Michael HO O O HO O Aldol O O Aldol HO O O HO Michael O O O Aldol There are other possibilities for order. O O HO Aldol O O 5.) Synthesize the following molecules. All carbons in the molecules must come from benzene or compounds with 5C’s or less. a.) O H2SO4 O HNO3 O2N AlCl3 O O SOCl2 HO Cl H2CrO4 1.) BuLi, Et2O + O 2.) H3O HO b.) O O O Cl + H3O NaOEt, EtOH O O O O Cl O AlCl3 Cl Cl AlCl3 Cl2 c.) O OMe NaOMe MeOH O O 1.) POCl3, DMF 2.) H2O OMe OMe O AlCl3 MeI Cl ONa HCl NaOH ZnHg 1.) NaOH + mcpba O 2.) H3O O OH O O AlCl3 Cl 6.) Write the mechanism for the following reactions. -

Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— a Literature Review
Molecules 2013, 18, 11873-11903; doi:10.3390/molecules181011873 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— A Literature Review Sowmini Kumaran 1, Mulchand S. Patel 2 and Frank Jordan 1,* 1 Department of Chemistry, Rutgers University, Newark, NJ 07102, USA 2 Department of Biochemistry, School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14214, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-973-353-5470; Fax: +1-973-353-1264. Received: 30 July 2013; in revised form: 14 September 2013 / Accepted: 16 September 2013 / Published: 26 September 2013 Abstract: The 2-oxoacid dehydrogenase complexes (ODHc) consist of multiple copies of three enzyme components: E1, a 2-oxoacid decarboxylase; E2, dihydrolipoyl acyl-transferase; and E3, dihydrolipoyl dehydrogenase, that together catalyze the oxidative decarboxylation of 2-oxoacids, in the presence of thiamin diphosphate (ThDP), coenzyme A 2+ + (CoA), Mg and NAD , to generate CO2, NADH and the corresponding acyl-CoA. The structural scaffold of the complex is provided by E2, with E1 and E3 bound around the periphery. The three principal members of the family are pyruvate dehydrogenase (PDHc), 2-oxoglutarate dehydrogenase (OGDHc) and branched-chain 2-oxo acid dehydrogenase (BCKDHc). In this review, we report application of NMR-based approaches to both mechanistic and structural issues concerning these complexes. These studies revealed the nature and reactivity of transient intermediates on the enzymatic pathway and provided site-specific information on the architecture and binding specificity of the domain interfaces using solubilized truncated domain constructs of the multi-domain E2 component in its interactions with the E1 and E3 components. -

Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids
International Journal of Molecular Sciences Article Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids Stefan R. Marsden, Duncan G. G. McMillan and Ulf Hanefeld * Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Van der Maasweg 9, 2629HZ Delft, The Netherlands; [email protected] (S.R.M.); [email protected] (D.G.G.M.) * Correspondence: [email protected] Received: 12 October 2020; Accepted: 12 November 2020; Published: 16 November 2020 Abstract: The synthetic properties of the Thiamine diphosphate (ThDP)-dependent pyruvate dehydrogenase E1 subunit from Escherichia coli (EcPDH E1) was assessed for carboligation reactions with aliphatic ketoacids. Due to its role in metabolism, EcPDH E1 was previously characterised with respect to its biochemical properties, but it was never applied for synthetic purposes. Here, we show that EcPDH E1 is a promising biocatalyst for the production of chiral α-hydroxyketones. WT EcPDH E1 shows a 180–250-fold higher catalytic efficiency towards 2-oxobutyrate or pyruvate, respectively, in comparison to engineered transketolase variants from Geobacillus stearothermophilus (TKGST). Its broad active site cleft allows for the efficient conversion of both (R)- and (S)-configured α-hydroxyaldehydes, next to linear and branched aliphatic aldehydes as acceptor substrates under kinetically controlled conditions. The alternate, thermodynamically controlled self-reaction of aliphatic aldehydes was shown to be limited to low levels of conversion, which we propose to be due to their large hydration constants. Additionally, the thermodynamically controlled approach was demonstrated to suffer from a loss of stereoselectivity, which makes it unfeasible for aliphatic substrates. Keywords: Thiamine diphosphate; transketolase; C-C bond formation; kinetic control; acyloins 1. -
![Umpolung of Amine Reactivity. Nucleophilic [Alpha]](https://docslib.b-cdn.net/cover/9062/umpolung-of-amine-reactivity-nucleophilic-alpha-2689062.webp)
Umpolung of Amine Reactivity. Nucleophilic [Alpha]
11591 S. W. Benson: Thermochemical Kinetics. Wiley, New York, N. Y., [I631 C. M. Shy and J. F. Finklea, Environ. Sci. Technol. 7, 205 (1973). 1968. [I641 R. P. Steer, K. R. Darnall, and J. N. Pifrs, Jr., Tetrahedron Lett. [I601 J. G. Caluert, K. L. Demerjian, and J. A. Kerr, Proc. Int. Symp. 1969. 3765. Air Pollut., Tokyo, Oct. 17-19, 1972, pp. 465ff. 11651 S. Furuyama. R. Atkinson, A. J. Colussi, and R. J. Cvetanouic, Int. [161] A. Q. Eschenroeder and J. R. Marfinez, Advan. Chem. Ser. 113, 101 J. Chem. Kinet. 6, 741 (1974). (1972). [166] J. N. Pitrs, Jr., P. G. Bekowies. G. J. Doyle, J. M. McAfee, and [I621 7: A. Hecht, J. H. Seinfeld. and M. C. Dodge, Environ. Sci. Technol. A. M. Winer, to be oublished. 8, 327 (1974). and references therein. Umpolung of Amine Reactivity. Nucleophilic a-(Secondary Amino)- New synthetic alky lation via Metalated Nitrosaminesr**lr”’l methods 0 By Dieter Seebach and Dieter Ended*] There are basically two kinds of hetero atoms in organic molecules: one kind confers electrophilic character upon the carbon atom to which it is bound, and the other kind turns it into a nucleophilic site. The development of methods permitting transitions between the two resulting categories of reagents has become an important task of modern organic synthesis. The scope of such umpolung of the reactivity of functional groups is discussed for the case of amines as an example. A method of preparing masked u-secondary amino carbanions consists in nitrosation of the secondary amine, followed by metalation of the resulting nitrosamine CL to the nitrogen, reaction with electrophiles, and subsequent denitrosation. -

Organic Chemistry 2 Ester Enolates and Acyl Anion Equivalents
Dr. Peter Wipf Chemistry 0320 - Organic Chemistry 2 Ester Enolates and Acyl Anion Equivalents The Claisen condensation is the ester analog of the aldol reaction. Under standard conditions (sodium alkoxide, alkohol), the equilibrium is shifted to the β-dicarbonyl compound as a consequence of the irreversible enolization to the highly resonance-stabilized dicarbonyl enolate. O O NaOR, HOR O O O O + OR R' R' R' R'=OR, C The intramolecular Claisen reaction is generally called the Dieckmann condensation. Another modification of the Claisen condensation is the use of strong base which leads to irreversible enolization of the carbonyl compounds. O OLi LDA, -78 °C PhCOCl O O OEt OEt THF Ph OEt β-Dicarbonyl, and, generally, active methylene compounds provide a rich source of chemistry in alkylation reactions, due to their relative ease of enolization and the possibility for removal of one of the activating groups. Z1 Z2 , Z = ester, ketone, amide, nitrile, aldehyde, nitro, sulfoxide, sulfone, sulfonate, sulfonamide H H Especially important are acetoacetate and malonate building blocks (acetoacetic and malonic ester syntheses): O O O O O O OEt OEt OEt XCH2CO2R R-X R-X (1 equiv) RCOX O O O O OH O O R R OEt O R Dithioketals can be used in Umpolung reactions as latent carbonyl groups. Nature’s Umpolung reagent is thiamine pyrophosphate (TPP). In the pentose phosphate pathway, TPP catalyzes the conversion of erythrose into fructose. First, the carbanion of TPP attacks the carbonyl carbon of xylulose 5-phosphate, forming a proton from the TPP adduct, and subsequent electron rearrangement leads to fragmentation, releasing the first product, glyceraldehyde 3-phosphate. -
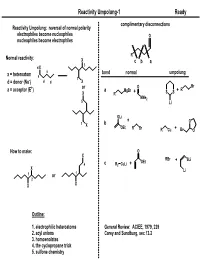
Reactivity Umpolung-1
Reactivity Umpolung-1 Ready complimentary disconnections Reactivity Umpolung: reversal of normal polarity electrophiles become nucleophiles O nucleophiles become electrophiles R Normal reactivity: X c ba 3 d X a bond normal umpolung x = heteroatom a 1 d = donor (Nu-) d d X + or O Br a = acceptor (E ) a MgBr + R X R + SS NMe2 5 Li OLi + O 1 b X OEt R Br R Cu + Br O How to make: O X RBr OEt + SLi 4 c R2 CuLi + X Li 1 or 1 2 X X Outline: 1. electrophilic heteroatoms General Review: ACIEE, 1979, 239 2. acyl anions Carey and Sundburg, sec 13.2 3. homoenolates 4. the cyclopropane trick 5. sulfone chemistry Reactivity Umpolung-2 Ready Electrophilic Heteroatoms mCPBA 'Rubottom [O]' OTMS 1. mCPBA OTMS O General form: 2. HF Ph Ph Ph 60% O OH X X Y+ X,Y = heteroatoms Y Rubottom, TL, 1974, 4319 Electrophile Examples O O O O2 O O 1. LDA; O2 O 1. TBSOTf, Et3N DMDO PO PO OTBS NMe2 2. Na2S2O3 HO 2. DMDO NMe2 84% P'O OP" 69% P'O OP" OP" O OP" HO O NMe2 Falck, JOC, 1995, 3385 Oxaziridines 1. LiHMDS 2. " HO Ph CO2Et Ph CO2Et 81% O O N OH O S O Wasserman and Lipshutz O TL, 1975, 1731 O 95% ee O O Mo O O Davis, Chem. Rev. 1992, 919 O O OH Py O LDA; MoOPH not a general asymmetric method. For racemic, most 87% commonly used is 'Davis Oxaziridine' P(NMe2)3 O MoOPH Ph Vedejs, JOC, 1978, 188 TsN Reactivity Umpolung-3 Ready Aminations (Review: Syn Lett, 1997, 741) Heteroatom exchange: Most commonly with Br, Cl Electrophile Examples O O cat PCl3 O NH3 R R R OH E OH OH Br2 OH N 1. -

Methods of Reactivity Umpolung
Volume 18 - Number 4 April 1979 Pages 239 - 336 International Edition in English Methods of Reactivity Umpolung By Dieter SeebachL"] Dedicated to Professor Horst Pommer on the occasion of his 60th birthday The past decade of organic chemistry may be characterized as a period of violent development of new synthetic methods. This was accompanied by a systematization of the analysis of synthetic problems (synthetic strategy). The planning of the synthesis of an organic target molecule is greatly facilitated by distinguishing between reagents X(C),, . with normal reactiv- ity (acceptor properties at C'*3,5-, donor properties at X, C2,4-.)and with reactivity umpolung (acceptor properties at X, C2,"...,donorproperties at C',3,5-.).In this context, reactivity umpolung turned out to be useful as a heuristic principle, as a classification scheme, and as an aid for locating so-called strategic bonds (synthon, transform, and antithesis according to E. J. Corey). There are six principal methods of umpolung: 1,2n-oxidation, heteroatom exchange and modification, homologation and its reversal, the cyclopropane "trick", use of acetylenes, and redox reactions; under certain circumstances none of these techniques is necessary in cases where direct umpolung is possible. Throughout the article, normal reactivity is indicated by green print; reactivity umpolung by red print. 1. Postulates and Nomenclature 3. These heteroatoms impose an alternating acceptor and donor reactivity pattern (I upon the carbon skeleton, i.e. Before describing and systematizing the methods of reactiv- acceptor properties or attack by donors at carbons C'.3.5-, ity umpolung, the scope of the discussion, the nomenclature and donor properties or attack by acceptors at carbons of synthetic methodology and the synthetic problem behind C2.4.6.-;the heteroatom Xo itself is a donor center tl"['al. -

Photocatalytic Umpolung Synthesis of Nucleophilic Π-Allylcobalt
pubs.acs.org/acscatalysis Research Article Photocatalytic Umpolung Synthesis of Nucleophilic π‑Allylcobalt Complexes for Allylation of Aldehydes Caizhe Shi, Fusheng Li, Yuqing Chen, Shuangjie Lin, Erjun Hao, Zhuowen Guo, Urwa Tul Wosqa, Dandan Zhang, and Lei Shi* Cite This: ACS Catal. 2021, 11, 2992−2998 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: The concept of “umpolung” reactivity of π-allylmetal complexes has been developed as a powerful method for the allylation of aldehydes. This paper describes the photocatalytic umpolung strategy for the synthesis of nucleophilic allylcobalt complexes through a single-electron-transfer (SET) process. This strategy enables the metallaphotoredox allylation of carbonyls with allyl acetate using organic N,N-diisopropylethylamine as the terminal reductant bypassing the use of a stoichiometric amount of metals. Ultraviolet−visible spectroscopy was used to monitor the redox changes of cobalt in the reaction. KEYWORDS: π-allylcobalt complexes, photoredox catalysis, allylation, umpolung, alcohol omoallylic alcohols and their derivatives are essential Metallaphotoredox catalysis is a new and rapidly growing − H synthetic intermediates for the preparation of numerous research field.17 25 Recently, the combination of photoredox natural products and medicine.1,2 The concept of “umpolung” and cobalt catalysis has been developed as a distinct new option reactivity of π-allylmetal complexes has been developed as a in the field of metallaphotoredox catalysis and has found powerful approach for carbonyl allylation with high regio- and 26−44 − numerous applications in synthetic chemistry. Given that diastereo-selectivity control (Scheme 1a).3 5 Such “reverse photoredox processes can directly modify the oxidation state of reactivity” of π-allylmetal complexes can be controlled by the transition metals by single electron transfer (SET), we wonder modification of their electronic environment through the use of whether the photoredox system can convert an electrophilic π- additives and ligands. -

Driving Recursive Dehydration by PIII/PV Catalysis: Annulation of Amines and Carboxylic Acids by Sequential C–N and C–C Bond Formation
Driving Recursive Dehydration by PIII/PV Catalysis: Annulation of Amines and Carboxylic Acids by Sequential C–N and C–C Bond Formation The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Lecomte, Morgan, Jeffrey M. Lipshultz, Shin-Ho Kim-Lee, Gen Li, and Alexander T. Radosevich, "Driving Recursive Dehydration by PIII/PV Catalysis: Annulation of Amines and Carboxylic Acids by Sequential C–N and C–C Bond Formation." Journal of the American Chemical Society 141, 32 (July 2019): p. 12507-12 doi 10.1021/ jacs.9b06277 ©2019 Author(s) As Published 10.1021/jacs.9b06277 Publisher American Chemical Society (ACS) Version Final published version Citable link https://hdl.handle.net/1721.1/124882 Terms of Use Creative Commons Attribution-NonCommercial-NoDerivs License Detailed Terms http://creativecommons.org/licenses/by-nc-nd/4.0/ This is an open access article published under a Creative Commons Non-Commercial No Derivative Works (CC-BY-NC-ND) Attribution License, which permits copying and redistribution of the article, and creation of adaptations, all for non-commercial purposes. Communication Cite This: J. Am. Chem. Soc. 2019, 141, 12507−12512 pubs.acs.org/JACS Driving Recursive Dehydration by PIII/PV Catalysis: Annulation of Amines and Carboxylic Acids by Sequential C−N and C−C Bond Formation † § † § † ‡ † Morgan Lecomte, , Jeffrey M. Lipshultz, , Shin-Ho Kim-Lee, , Gen Li, † and Alexander T. Radosevich*, † Department of Chemistry, Massachusetts Institute of Technology, 02139 Cambridge, Massachusetts, United States ‡ Departamento de Química Organica,́ Facultad de Ciencias, Universidad Autonomá de Madrid, Cantoblanco, 28049 Madrid, Spain *S Supporting Information ABSTRACT: A method for the annulation of amines and carboxylic acids to form pharmaceutically relevant azaheterocycles via organophosphorus PIII/PV redox catalysis is reported. -

Umpolung Strategy: Advances in Catalytic C-C Bond Formations
Turkish Journal of Chemistry Turk J Chem (2013) 37: 586 { 609 http://journals.tubitak.gov.tr/chem/ ⃝c TUB¨ ITAK_ Research Article doi:10.3906/kim-1303-85 Umpolung strategy: advances in catalytic C-C bond formations Serkan EYMUR,1;2 Mehmet GOLL¨ U,¨ 1 Cihangir TANYELI_1∗ 1Department of Chemistry, Middle East Technical University, Ankara, Turkey 2Department of Chemistry, Sel¸cukUniversity, Konya, Turkey Received: 27.03.2013 • Accepted: 28.04.2013 • Published Online: 12.07.2013 • Printed: 05.08.2013 Abstract: This mini-review examines the recent advances in umpolung strategy, devised originally by Corey and Seebach. Although numerous stoichiometric variants have been published to date, this article covers just the catalytic C-C bond forming reactions due to their major benefits such as atom economy, less pollution, and workable simplicity. In the context of umpolung, the studies are evaluated under 3 main titles: enzyme, N -heterocyclic carbene, and cyanide ion catalyzed reactions. In particular, enzyme and NHC catalyzed reactions mainly involve asymmetric applications. Key words: Umpolung, thiamine pyrophosphate (TPP), N -heterocyclic carbene, asymmetric synthesis 1. Introduction to umpolung As an outcome of extensive efforts dedicated to the development of new approaches for the construction of either complex synthetic targets or small synthetic building blocks, numerous synthetic organic methods are now available for a wide range of bond-forming reactions. 1 In particular, carbon-carbon (C-C) bond-forming reactions have been the key point among all the organic reactions in the history of organic synthesis. 2 However, the investigation of catalytic methods for C-C bond formation reactions, while generating functionality, remains a formidable challenge in the ongoing development of effective and reliable chemical processes. -

Imino Esters
DOI 10.1515/pac-2013-1105 Pure Appl. Chem. 2014; 86(5): 755–764 Conference paper Kaichiro Koyama, Isao Mizota and Makoto Shimizu* Integrated reactions based on the sequential addition to α-imino esters Abstract: This article summarizes integrated sequential reactions with α-imino esters, where the umpolung addition reaction to the imino nitrogen followed by the second addition or oxidation is the crucial step. The following four types of reactions are discussed: (1) tandem N-ethylation/Mannich reaction; (2) N-alkylation/ addition reaction; (3) synthesis of indolin-3-ones and tetrahydro-4-quinolones; (4) regioselective tandem N-alkylation/ C-acylation of β,γ-alkynyl α-imino esters. Keywords: alkoxycarbonyl iminium salt; α-imino ester; indolin-3-one; N-alkylation; NMS-IX; tetrahydro- 4-quinolone; umpolung. *Corresponding author: Makoto Shimizu, Department of Chemistry for Materials, Mie University, Tsu, Mie 514-8507, Japan, e-mail: [email protected] Kaichiro Koyama and Isao Mizota: Department of Chemistry for Materials, Mie University, Tsu, Mie 514-8507, Japan Article note: Paper based on a presentation at the 9th International Symposium on Novel Materials and their Synthesis (NMS-IX) and the 23rd International Symposium on Fine Chemistry and Functional Polymers (FCFP-XXIII), Shanghai, China, 17–22 October 2013. This paper is dedicated to the memory of Prof. Yingyan Jiang. Introduction Synthetic methods for the preparation of nitrogen-containing esters are of utmost interest and importance because these structures are the key components of natural and unnatural biologically active compounds and functionalized materials. The most straightforward approach to synthesize amino esters involves chemoselective nucleophilic additions to the imino groups of imino esters, and many examples have been reported in which various nucleophiles add to imines in a 1,2-fashion.