Modern Organic Synthesis an Introduction Chapter 1 Synthetic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Philosophy of Science and Philosophy of Chemistry
Philosophy of Science and Philosophy of Chemistry Jaap van Brakel Abstract: In this paper I assess the relation between philosophy of chemistry and (general) philosophy of science, focusing on those themes in the philoso- phy of chemistry that may bring about major revisions or extensions of cur- rent philosophy of science. Three themes can claim to make a unique contri- bution to philosophy of science: first, the variety of materials in the (natural and artificial) world; second, extending the world by making new stuff; and, third, specific features of the relations between chemistry and physics. Keywords : philosophy of science, philosophy of chemistry, interdiscourse relations, making stuff, variety of substances . 1. Introduction Chemistry is unique and distinguishes itself from all other sciences, with respect to three broad issues: • A (variety of) stuff perspective, requiring conceptual analysis of the notion of stuff or material (Sections 4 and 5). • A making stuff perspective: the transformation of stuff by chemical reaction or phase transition (Section 6). • The pivotal role of the relations between chemistry and physics in connection with the question how everything fits together (Section 7). All themes in the philosophy of chemistry can be classified in one of these three clusters or make contributions to general philosophy of science that, as yet , are not particularly different from similar contributions from other sci- ences (Section 3). I do not exclude the possibility of there being more than three clusters of philosophical issues unique to philosophy of chemistry, but I am not aware of any as yet. Moreover, highlighting the issues discussed in Sections 5-7 does not mean that issues reviewed in Section 3 are less im- portant in revising the philosophy of science. -

UMPOLUNG in REACTIONS CATALYZED by THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung En Reacciones Catalizadas Por Enzimas Dependientes De Pirofosfato De Tiamina
Ciencia, Ambiente y Clima, Vol. 2, No. 2, julio-diciembre, 2019 • ISSN (impreso): 2636-2317 • ISSN (en línea): 2636-2333 DOI: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 UMPOLUNG IN REACTIONS CATALYZED BY THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung en reacciones catalizadas por enzimas dependientes de pirofosfato de tiamina Carlos José Boluda Emily Soto Instituto Tecnológico de Santo Domingo (INTEC), Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales, Av. de Los Área de Ciencias Básicas y Ambientales Próceres 49, Santo Domingo, República Dominicana Correo-e: [email protected] *Corresponding author: Carlos J. Boluda Darah de la Cruz Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Carolina Juncá Área de Ciencias Básicas y Ambientales Instituto Tecnológico de Santo Domingo (INTEC), Correo-e: [email protected] Área de Ciencias Básicas y Ambientales Anny Peña Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales Correo-e: [email protected] Recibido: 25/9/2019 • Aprobado: 19/10/2019 Cómo citar: Boluda, C. J., Juncá, C., Soto, E., de la Cruz, D., & Peña, A. (2019). Umpolung in reactions catalyzed by thiamine pyrophos- phate dependent enzymes. Ciencia, Ambiente Y Clima, 2(2), 27-42. Doi: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 Abstract Resumen The temporal exchange of the electrophilic/nucleophilic El intercambio temporal del carácter electrofílico/nucleofí- character of an atom by chemical manipulation is known lico de un átomo mediante manipulación química, es cono- in organic chemistry as umpolung. This inversion of polarity cido con el vocablo alemán de umpolung. -

Modern Organic Synthesis an Introduction
Modern Organic Synthesis an Introduction G. S. Zweifel M. H. Nantz W.H. Freeman and Company Chapter 1 Synthetic Design • What is an ideal or viable synthesis, and how does one approach a synthetic project? • The overriding concern in a synthesis is the yield, including the inherent concepts of simplicity (fewest steps) and selectivity (chemoselectivity, regioselectivity, diastereoselectivity, and enantioselectivity). • This chapter outlines strategies for the synthesis of target molecules based on retrosynthetic analysis. 1 1.1 Retrosynthetic Analysis Basic Concept The symbol signifies a reverse synthetic step and is called atransform. The main transforms are disconnections, or cleavage of C-C bonds, and functional group interconversions (FGI) Retrosynthetic analysis involves the disassembly of a TM into available starting materials by sequential disconnections and functional group interconversions(FGI). Synthons are fragments resulting from disconnection of carbon-carbon bonds of the TM. The actual substrates used for the forward synthesis are the synthetic equivalents (SE). Synthetic design involves two distinct steps (1) Retrosynthetic analysis (2) Subsequent translation of the analysis into a “forward direction” synthesis. Chemical bonds can be cleaved heterolytically, homolytically, or through concerted transform. 2 Donor and Acceptor Synthons Acceptor synthon Æ carbocation (electrophilic) Donor synthon Æ carbanion (nucleophilic) Table 1.1 Common Acceptor Synthon Synthetic equivalents Common Acceptor Synthon Synthetic equivalents 3 Table 1.2 Common Donor Synthons Common Donor Synthon Synthetic equivalents Retrosynthetic Analysis A Synthesis A 4 Retrosynthetic Analysis B Synthesis B Alternating Polarity Disconnections The presence of a heteroatom in a molecule imparts a pattern of electrophilicity and nucleophilicity to the atom of the molecule. The concept of alternating polarities or latent polarities (imaginary chargies) often enables one to identify the best positions to make a disconnection within a complex molecule. -

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

Protokoll Handledarmöte 2020-02-14
http://www.diva-portal.org Postprint This is the accepted version of a paper published in Organic Letters. This paper has been peer-reviewed but does not include the final publisher proof-corrections or journal pagination. Citation for the original published paper (version of record): Colas, K., dos Santos, A C., Mendoza, A. (2019) i-Pr2-NMgCl·LiCl Enables the Synthesis of Ketones by Direct Addition of Grignard Reagents to Carboxylate Anions Organic Letters, 21(19): 7908-7913 https://doi.org/10.1021/acs.orglett.9b02899 Access to the published version may require subscription. N.B. When citing this work, cite the original published paper. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:su:diva-175818 i-Pr2NMgCl·LiCl Enables the Synthesis of Ketones by Direct Addi- tion of Grignard Reagents to Carboxylate Anions Kilian Colas, A. Catarina V. D. dos Santos and Abraham Mendoza* Department of Organic Chemistry, Stockholm University, Arrhenius Laboratory, 106 91 Stockholm (Sweden) Supporting Information Placeholder direct Grignard - carboxylate coupling 1 R MgX + i-Pr2NMgCl•LiCl 1 MgX ( ) R * CO2 i-Pr i-Pr L L O [Mg] N O ideal ( ) * Mg Li (*) sources 1 1 2 R O Cl R R R2 L O ketone [proposed structure] products base R1 OH [in-situ from commercial] ◾ >30 examples ◾ scalable ◾ facile isotopic-labelling ABSTRACT: The direct preparation of ketones from carboxylate anions is greatly limited by the required use of organolithium reagents or activated acyl sources that need to be independently prepared. Herein, a specific magnesium amide additive is used to activate and control the addition of more tolerant Grignard reagents to carboxylate anions. -
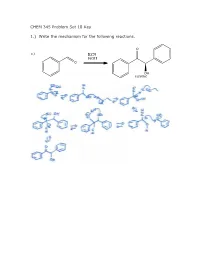
CHEM 345 Problem Set 18 Key 1.) Write the Mechanism for The
CHEM 345 Problem Set 18 Key 1.) Write the mechanism for the following reactions. O a.) KCN EtOH O OH racemic 1.) Write the mechanism for the following reactions. b.) KCN AcOH O NC OH racemic O c.) N S R O NEt3 OH racemic 2.) What is the structure of AcOH?Why does changing the solvent from EtOH to AcOH make such a big difference? O OH AcOH acetic acid The pKa of acetic acid is approximately 5. The pKa of ethanol is approximately 15. When you take a proton off of ethanol, you generate ethoxide which is about 1010 times stronger of a base than acetate. 3.) Give two instances when you need to use the thiazolium salt and triethylamine rather than KCN and EtOH. If the aldehydes contain an enolizable proton then you cannot use KCN/EtOH, instead you must use the thiazolium. Also, if the electrophile is a Michael acceptor to give a 1,4 dicarbonyl, then the thiazolium catalyst should be used. 4.) Break the following compound down as far as you can using Aldol, Michael, and Claisen reactions. Above each retrosynthetic arrow, write the name of the reaction. O HO O HO Aldol O Michael HO O O HO O Aldol O O Aldol HO O O HO Michael O O O Aldol There are other possibilities for order. O O HO Aldol O O 5.) Synthesize the following molecules. All carbons in the molecules must come from benzene or compounds with 5C’s or less. a.) O H2SO4 O HNO3 O2N AlCl3 O O SOCl2 HO Cl H2CrO4 1.) BuLi, Et2O + O 2.) H3O HO b.) O O O Cl + H3O NaOEt, EtOH O O O O Cl O AlCl3 Cl Cl AlCl3 Cl2 c.) O OMe NaOMe MeOH O O 1.) POCl3, DMF 2.) H2O OMe OMe O AlCl3 MeI Cl ONa HCl NaOH ZnHg 1.) NaOH + mcpba O 2.) H3O O OH O O AlCl3 Cl 6.) Write the mechanism for the following reactions. -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

Peptide Chemistry up to Its Present State
Appendix In this Appendix biographical sketches are compiled of many scientists who have made notable contributions to the development of peptide chemistry up to its present state. We have tried to consider names mainly connected with important events during the earlier periods of peptide history, but could not include all authors mentioned in the text of this book. This is particularly true for the more recent decades when the number of peptide chemists and biologists increased to such an extent that their enumeration would have gone beyond the scope of this Appendix. 250 Appendix Plate 8. Emil Abderhalden (1877-1950), Photo Plate 9. S. Akabori Leopoldina, Halle J Plate 10. Ernst Bayer Plate 11. Karel Blaha (1926-1988) Appendix 251 Plate 12. Max Brenner Plate 13. Hans Brockmann (1903-1988) Plate 14. Victor Bruckner (1900- 1980) Plate 15. Pehr V. Edman (1916- 1977) 252 Appendix Plate 16. Lyman C. Craig (1906-1974) Plate 17. Vittorio Erspamer Plate 18. Joseph S. Fruton, Biochemist and Historian Appendix 253 Plate 19. Rolf Geiger (1923-1988) Plate 20. Wolfgang Konig Plate 21. Dorothy Hodgkins Plate. 22. Franz Hofmeister (1850-1922), (Fischer, biograph. Lexikon) 254 Appendix Plate 23. The picture shows the late Professor 1.E. Jorpes (r.j and Professor V. Mutt during their favorite pastime in the archipelago on the Baltic near Stockholm Plate 24. Ephraim Katchalski (Katzir) Plate 25. Abraham Patchornik Appendix 255 Plate 26. P.G. Katsoyannis Plate 27. George W. Kenner (1922-1978) Plate 28. Edger Lederer (1908- 1988) Plate 29. Hennann Leuchs (1879-1945) 256 Appendix Plate 30. Choh Hao Li (1913-1987) Plate 31. -

Synthesis Infographic (Revised)
Organic Synthesis A problem–solving guide for Organic Chemistry I/II ? Goal: propose a synthesis of a target molecule H OH N using given starting materials and any other N reagents you need. starting target materials molecule These key strategies should guide your approach:1 Mapping • Systematically label all the atoms in the ? 1 starting materials, then identify those atoms in H OH N 6 N the target molecule 2 4 5 7 6 1 3 5 7 2 3 4 • Use landmarks (heteroatoms, functional groups) to help you Identifying bonds formed/broken ? and atoms added/removed H OH 1 N 6 N Having labeled atoms, it becomes easier to see: 2 4 5 7 6 1 3 5 7 2 3 4 bonds formed: C7–N2 σ, C6–O σ atoms added • where bonds have been formed/broken bonds broken: C6–C7 π atoms removed • where atoms (including protons) have been added/removed ? Regiochemical and stereochemical analysis H OH 1 N 6 N 2 4 5 7 6 • Look for regiochemical patterns that can 1 3 5 7 2 3 4 provide ideas for synthetic strategies target molecule is 1,2–difunctionalized at C6–C7 • Look for changes in configuration at C7–N2 bond formed at less substituted C of propylene stereocenters (not always applicable) reaction: base–catalyzed epoxide opening 1 OH OH 1 Synthon-based retrosynthetic analysis N + N 6 2 4 6 2 4 Construct hypothetical starting materials for 5 7 3 5 7 3 • synthons forming required bonds 1 This process is called constructing synthons, O δ- • + + HN 6 6 δ 2 4 and it is the basis of retrosynthetic analysis 5 7 5 7 3 possible reagents • Synthons are a tool for choosing reagents 1Based in part on strategies observed in successful students’ responses to synthetic Flynn Research Group 2017 – FlynnResearchGroup.com problems on exams; see: Bodé, N. -

Bsc Chemistry
__________________________________________________________________________________________________ Subject Chemistry Paper No and Title 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No and 2: Synthons, Synthetic equivalents and Title Retrosynthetic analysis Module Tag CHE_P14_M2 CHEMISTRY Paper No. 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No. 2: Synthons, Synthetic equivalents and Retrosynthetic analysis __________________________________________________________________________________________________ TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. Synthons and Synthetic equivalents 4. Retrosynthetic analysis 5. Summary CHEMISTRY Paper No. 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No. 2: Synthons, Synthetic equivalents and Retrosynthetic analysis __________________________________________________________________________________________________ 1. Learning Outcomes After studying this module, you shall be able to: Know about synthons Know about synthetic equivalents Know about retrosynthetic analysis. Study the steps involved in retrosynthetic analysis. Know the routine for designing the synthesis. 2. Introduction Organic chemistry is a branch of chemistry which deals with the study of carbon and its compounds. The study includes the understanding of synthesis, structure, properties, and reactions of organic compounds. -

Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— a Literature Review
Molecules 2013, 18, 11873-11903; doi:10.3390/molecules181011873 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— A Literature Review Sowmini Kumaran 1, Mulchand S. Patel 2 and Frank Jordan 1,* 1 Department of Chemistry, Rutgers University, Newark, NJ 07102, USA 2 Department of Biochemistry, School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14214, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-973-353-5470; Fax: +1-973-353-1264. Received: 30 July 2013; in revised form: 14 September 2013 / Accepted: 16 September 2013 / Published: 26 September 2013 Abstract: The 2-oxoacid dehydrogenase complexes (ODHc) consist of multiple copies of three enzyme components: E1, a 2-oxoacid decarboxylase; E2, dihydrolipoyl acyl-transferase; and E3, dihydrolipoyl dehydrogenase, that together catalyze the oxidative decarboxylation of 2-oxoacids, in the presence of thiamin diphosphate (ThDP), coenzyme A 2+ + (CoA), Mg and NAD , to generate CO2, NADH and the corresponding acyl-CoA. The structural scaffold of the complex is provided by E2, with E1 and E3 bound around the periphery. The three principal members of the family are pyruvate dehydrogenase (PDHc), 2-oxoglutarate dehydrogenase (OGDHc) and branched-chain 2-oxo acid dehydrogenase (BCKDHc). In this review, we report application of NMR-based approaches to both mechanistic and structural issues concerning these complexes. These studies revealed the nature and reactivity of transient intermediates on the enzymatic pathway and provided site-specific information on the architecture and binding specificity of the domain interfaces using solubilized truncated domain constructs of the multi-domain E2 component in its interactions with the E1 and E3 components. -

Recoverable Phospha-Michael Additions Catalyzed by a 4-N,N
molecules Article Recoverable Phospha-Michael Additions Catalyzed by a 4-N,N-Dimethylaminopyridinium Saccharinate Salt or a Fluorous Long-Chained Pyridine: Two Types of Reusable Base Catalysts Eskedar Tessema 1,†, Vijayanath Elakkat 1,† , Chiao-Fan Chiu 2,3,*, Jing-Hung Zheng 1, Ka Long Chan 1, Chia-Rui Shen 4,5, Peng Zhang 6,* and Norman Lu 1,7,* 1 Institute of Organic and Polymeric Materials, National Taipei University of Technology, Taipei 106, Taiwan; [email protected] (E.T.); [email protected] (V.E.); [email protected] (J.-H.Z.); [email protected] (K.L.C.) 2 Department of Pediatrics, Linkou Medical Center, Chang Gung Memorial Hospital, Taoyuan 333, Taiwan 3 Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan 4 Department of Medical Biotechnology and Laboratory Sciences, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan; [email protected] 5 Department of Ophthalmology, Linkou Medical Center, Chang Gung Memorial Hospital, Taoyuan 333, Taiwan 6 Department of Chemistry, University of Cincinnati, Cincinnati, OH 45221-0172, USA 7 Development Center for Smart Textile, National Taipei University of Technology, Taipei 106, Taiwan Citation: Tessema, E.; Elakkat, V.; * Correspondence: [email protected] (C.-F.C.); [email protected] (P.Z.); Chiu, C.-F.; Zheng, J.-H.; Chan, K.L.; [email protected] (N.L.) Shen, C.-R.; Zhang, P.; Lu, N. † These authors contributed equally to this work. Recoverable Phospha-Michael Additions Catalyzed by a Abstract: Phospha-Michael addition, which is the addition reaction of a phosphorus-based nucle- 4-N,N-Dimethylaminopyridinium ophile to an acceptor-substituted unsaturated bond, certainly represents one of the most versatile and Saccharinate Salt or a Fluorous powerful tools for the formation of P-C bonds, since many different electrophiles and P nucleophiles Long-Chained Pyridine: Two Types can be combined with each other.