Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UMPOLUNG in REACTIONS CATALYZED by THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung En Reacciones Catalizadas Por Enzimas Dependientes De Pirofosfato De Tiamina
Ciencia, Ambiente y Clima, Vol. 2, No. 2, julio-diciembre, 2019 • ISSN (impreso): 2636-2317 • ISSN (en línea): 2636-2333 DOI: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 UMPOLUNG IN REACTIONS CATALYZED BY THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung en reacciones catalizadas por enzimas dependientes de pirofosfato de tiamina Carlos José Boluda Emily Soto Instituto Tecnológico de Santo Domingo (INTEC), Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales, Av. de Los Área de Ciencias Básicas y Ambientales Próceres 49, Santo Domingo, República Dominicana Correo-e: [email protected] *Corresponding author: Carlos J. Boluda Darah de la Cruz Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Carolina Juncá Área de Ciencias Básicas y Ambientales Instituto Tecnológico de Santo Domingo (INTEC), Correo-e: [email protected] Área de Ciencias Básicas y Ambientales Anny Peña Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales Correo-e: [email protected] Recibido: 25/9/2019 • Aprobado: 19/10/2019 Cómo citar: Boluda, C. J., Juncá, C., Soto, E., de la Cruz, D., & Peña, A. (2019). Umpolung in reactions catalyzed by thiamine pyrophos- phate dependent enzymes. Ciencia, Ambiente Y Clima, 2(2), 27-42. Doi: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 Abstract Resumen The temporal exchange of the electrophilic/nucleophilic El intercambio temporal del carácter electrofílico/nucleofí- character of an atom by chemical manipulation is known lico de un átomo mediante manipulación química, es cono- in organic chemistry as umpolung. This inversion of polarity cido con el vocablo alemán de umpolung. -

Flavin-Containing Monooxygenases: Mutations, Disease and Drug Response Phillips, IR; Shephard, EA
Flavin-containing monooxygenases: mutations, disease and drug response Phillips, IR; Shephard, EA For additional information about this publication click this link. http://qmro.qmul.ac.uk/jspui/handle/123456789/1015 Information about this research object was correct at the time of download; we occasionally make corrections to records, please therefore check the published record when citing. For more information contact [email protected] Flavin-containing monooxygenases: mutations, disease and drug response Ian R. Phillips1 and Elizabeth A. Shephard2 1School of Biological and Chemical Sciences, Queen Mary, University of London, Mile End Road, London E1 4NS, UK 2Department of Biochemistry and Molecular Biology, University College London, Gower Street, London WC1E 6BT, UK Corresponding author: Shephard, E.A. ([email protected]). and, thus, contribute to drug development. This review Flavin-containing monooxygenases (FMOs) metabolize considers the role of FMOs and their genetic variants in numerous foreign chemicals, including drugs, pesticides disease and drug response. and dietary components and, thus, mediate interactions between humans and their chemical environment. We Mechanism and structure describe the mechanism of action of FMOs and insights For catalysis FMOs require flavin adenine dinucleotide gained from the structure of yeast FMO. We then (FAD) as a prosthetic group, NADPH as a cofactor and concentrate on the three FMOs (FMOs 1, 2 and 3) that are molecular oxygen as a cosubstrate [5,6]. In contrast to most important for metabolism of foreign chemicals in CYPs FMOs accept reducing equivalents directly from humans, focusing on the role of the FMOs and their genetic NADPH and, thus, do not require accessory proteins. -
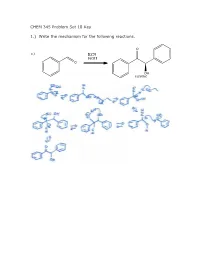
CHEM 345 Problem Set 18 Key 1.) Write the Mechanism for The
CHEM 345 Problem Set 18 Key 1.) Write the mechanism for the following reactions. O a.) KCN EtOH O OH racemic 1.) Write the mechanism for the following reactions. b.) KCN AcOH O NC OH racemic O c.) N S R O NEt3 OH racemic 2.) What is the structure of AcOH?Why does changing the solvent from EtOH to AcOH make such a big difference? O OH AcOH acetic acid The pKa of acetic acid is approximately 5. The pKa of ethanol is approximately 15. When you take a proton off of ethanol, you generate ethoxide which is about 1010 times stronger of a base than acetate. 3.) Give two instances when you need to use the thiazolium salt and triethylamine rather than KCN and EtOH. If the aldehydes contain an enolizable proton then you cannot use KCN/EtOH, instead you must use the thiazolium. Also, if the electrophile is a Michael acceptor to give a 1,4 dicarbonyl, then the thiazolium catalyst should be used. 4.) Break the following compound down as far as you can using Aldol, Michael, and Claisen reactions. Above each retrosynthetic arrow, write the name of the reaction. O HO O HO Aldol O Michael HO O O HO O Aldol O O Aldol HO O O HO Michael O O O Aldol There are other possibilities for order. O O HO Aldol O O 5.) Synthesize the following molecules. All carbons in the molecules must come from benzene or compounds with 5C’s or less. a.) O H2SO4 O HNO3 O2N AlCl3 O O SOCl2 HO Cl H2CrO4 1.) BuLi, Et2O + O 2.) H3O HO b.) O O O Cl + H3O NaOEt, EtOH O O O O Cl O AlCl3 Cl Cl AlCl3 Cl2 c.) O OMe NaOMe MeOH O O 1.) POCl3, DMF 2.) H2O OMe OMe O AlCl3 MeI Cl ONa HCl NaOH ZnHg 1.) NaOH + mcpba O 2.) H3O O OH O O AlCl3 Cl 6.) Write the mechanism for the following reactions. -

The Influence of Vitamin B12on the Content, Distribution and in Vivo
The Influence of Vitamin B12on the Content, Distribution and in Vivo Synthesis of Thiamine Pyrophosphate, Flavin Adenine Dinucleotide and Pyridine Nucleotides in Rat Liver1 URMILA MARFATIA, D. V. REGE, H. P. TIPNIS ANDA. SREENIVASAN Department of Chemical Technology, University of Bombay, Matunga, Bombay Apart from the well-known interrelation (FAD) and pyridine nucleotides (PN), and ships among the B vitamins, there is rea to their in vivo synthesis from the corre son to believe that folie acid and vitamin sponding administered vitamins, in the rat Downloaded from Bu may influence the functioning of other liver. Data on the distribution of these vitamins as cofactors. Thus, dietary folie cofactors in liver cells of the normal rat acid has been known to determine rat are available in the works of Goethart ('52) liver stores of coenzyme A (CoA) and and Dianzani and Dianzani Mor ('57) on adenotriphosphate (ATP) (Popp and Tot TPP, of Carruthers and Suntzeff ('54) and ter, '52; Totter, '53); a decrease in liver Dianzani ('55) on pyridine nucleotides DPN is also caused by aminopterin2 (PN) and of Schneider and Hogeboom jn.nutrition.org (Strength et al., '54). The in vivo incor (Schneider, '56) on FAD. poration of nitcotinamide into pyridino- nucleotides in rat liver is affected in a EXPERIMENTAL deficiency of vitamin B«(Nadkarni et al., Young, male Wistar rats weighing ap '57). Low blood level of citrovorum factor proximately 100 gm each were used. The by guest on April 19, 2011 in the hyperthyroid, vitamin Bi2-deficient animals, housed individually in raised rat is corrected by administration of vita mesh-bottom cages, were initially depleted min B«(Pfander et al., '52). -

Citric Acid Cycle
CHEM464 / Medh, J.D. The Citric Acid Cycle Citric Acid Cycle: Central Role in Catabolism • Stage II of catabolism involves the conversion of carbohydrates, fats and aminoacids into acetylCoA • In aerobic organisms, citric acid cycle makes up the final stage of catabolism when acetyl CoA is completely oxidized to CO2. • Also called Krebs cycle or tricarboxylic acid (TCA) cycle. • It is a central integrative pathway that harvests chemical energy from biological fuel in the form of electrons in NADH and FADH2 (oxidation is loss of electrons). • NADH and FADH2 transfer electrons via the electron transport chain to final electron acceptor, O2, to form H2O. Entry of Pyruvate into the TCA cycle • Pyruvate is formed in the cytosol as a product of glycolysis • For entry into the TCA cycle, it has to be converted to Acetyl CoA. • Oxidation of pyruvate to acetyl CoA is catalyzed by the pyruvate dehydrogenase complex in the mitochondria • Mitochondria consist of inner and outer membranes and the matrix • Enzymes of the PDH complex and the TCA cycle (except succinate dehydrogenase) are in the matrix • Pyruvate translocase is an antiporter present in the inner mitochondrial membrane that allows entry of a molecule of pyruvate in exchange for a hydroxide ion. 1 CHEM464 / Medh, J.D. The Citric Acid Cycle The Pyruvate Dehydrogenase (PDH) complex • The PDH complex consists of 3 enzymes. They are: pyruvate dehydrogenase (E1), Dihydrolipoyl transacetylase (E2) and dihydrolipoyl dehydrogenase (E3). • It has 5 cofactors: CoASH, NAD+, lipoamide, TPP and FAD. CoASH and NAD+ participate stoichiometrically in the reaction, the other 3 cofactors have catalytic functions. -

Roles of Vitamin Metabolizing Genes in Multidrug-Resistant Plasmids of Superbugs
bioRxiv preprint doi: https://doi.org/10.1101/285403; this version posted March 20, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Roles of Vitamin Metabolizing Genes in Multidrug-Resistant Plasmids of Superbugs Asit Kumar Chakraborty, Genetic Engineering Laboratory, Department of Biotechnology & Biochemistry, Oriental Institute of Science & Technology, Vidyasagar University, Midnapore 721102, West Bengal, India. Abstract Superbug crisis has rocked this world with million deaths due to failure of potent antibiotics. Thousands mdr genes with hundreds of mutant isomers are generated. Small integrons and R-plasmids have combined with F'-plasmids creating a space for >10-20 of mdr genes that inactivate antibiotics in different mechanisms. Mdr genes are created to save bacteria from antibiotics because gut microbiota synthesize >20 vitamins and complex bio-molecules needed for >30000 biochemical reactions of human metabolosome. In other words, mdr gene creation is protected from both sides, intestinal luminal cells and gut bacteria in a tight symbiotic signalling system. We have proposed, to avert the crisis all vitamin metabolizing genes will be acquired in MDR- plasmids if we continue oral antibiotics therapy. Therefore, we have checked the plasmid databases and have detected thiamine, riboflavin, folate, cobalamine and biotin metabolizing enzymes in MDR plasmids. Thus vit genes may mobilise recently into MDR-plasmids and are likely essential for gut microbiota protection. Analysis found that cob and thi genes are abundant and likely very essential than other vit genes. -

Cofactor Strap Regulates Oxidative Phosphorylation and Mitochondrial P53 Activity Through ATP Synthase
Cell Death and Differentiation (2015) 22, 156–163 & 2015 Macmillan Publishers Limited All rights reserved 1350-9047/15 www.nature.com/cdd Cofactor Strap regulates oxidative phosphorylation and mitochondrial p53 activity through ATP synthase S Maniam1,4, AS Coutts1,4, MR Stratford2, J McGouran3, B Kessler3 and NB La Thangue*,1 Metabolic reprogramming is a hallmark of cancer cells. Strap (stress-responsive activator of p300) is a novel TPR motif OB-fold protein that contributes to p53 transcriptional activation. We show here that, in addition to its established transcriptional role, Strap is localised at mitochondria where one of its key interaction partners is ATP synthase. Significantly, the interaction between Strap and ATP synthase downregulates mitochondrial ATP production. Under glucose-limiting conditions, cancer cells are sensitised by mitochondrial Strap to apoptosis, which is rescued by supplementing cells with an extracellular source of ATP. Furthermore, Strap augments the apoptotic effects of mitochondrial p53. These findings define Strap as a dual regulator of cellular reprogramming: first as a nuclear transcription cofactor and second in the direct regulation of mitochondrial respiration. Cell Death and Differentiation (2015) 22, 156–163; doi:10.1038/cdd.2014.135; published online 29 August 2014 An established characteristic of tumour cells is their under- model whereby Strap unfolds to become more accessible lying metabolic changes.1 Early observations that during the DNA-damage response.12 tumour cells had persistently high -

Coenzymes and Prosthetic Groups Nomenclature
Coenzymes and prosthetic groups Nomenclature • Cofactor: nonprotein component of enzymes • Cofactor - a co-catalyst required for enzyme activity • Coenzyme - a dissociable cofactor, usually organic • Prosthetic group - non-dissociable cofactor • Vitamin - a required micro-nutrient (organism cannot synthesize adequate quantities for normal health - may vary during life-cycle). – water soluble - not stored, generally no problem with overdose – lipid soluble - stored, often toxic with overdose. • Apoenzyme - enzyme lacking cofactor (inactive) • Holoenzyme - enzyme with cofactors (active) Vitamins are precursors of cofactors Why cofactors? Adenine Nucleotide Coenzymes All use the adenine nucleotide group solely for binding to the enzyme! • pyridine dinucleotides (NADH, NADPH) • flavin mono- and dinucleotides (FMN, FADH) • coenzyme A Nucleotide triphosphates • ATP hydrolysis – resonance stabilizes products – reactants cannot be resonance stabilized because of competition with adjacent bridging anhydrides – charge density greater on reactants than products Coenzyme A • Activation of acyl groups for transfer by nucleophilic attack • activation of the alpha- hydrogen of the acyl group for abstraction as a proton • Both these functions are mediated by the reactive -SH group on CoA, which forms thioesters Coenzyme A Nicotinic Acid/Nicotinamide Coenzymes • These coenzymes are two-electron carriers • They transfer hydride anion (H-) to and from substrates • Two important coenzymes in this class: • Nicotinamide adenine dinucleotide (NAD+) • Nicotinamide -

Novel Derivatives of Nicotinamide Adenine Dinucleotide (NAD) and Their Biological Evaluation Against NAD- Consuming Enzymes
Novel derivatives of nicotinamide adenine dinucleotide (NAD) and their biological evaluation against NAD- Consuming Enzymes Giulia Pergolizzi University of East Anglia School of Pharmacy Thesis submitted for the degree of Doctor of Philosophy July, 2012 © This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with the author and that use of any information derived there from must be in accordance with current UK Copyright Law. In addition, any quotation or extract must include full attribution. ABSTRACT Nicotinamide adenine dinucleotide (β-NAD+) is a primary metabolite involved in fundamental biological processes. Its molecular structure with characteristic functional groups, such as the quaternary nitrogen of the nicotinamide ring, and the two high- energy pyrophosphate and nicotinamide N-glycosidic bonds, allows it to undergo different reactions depending on the reactive moiety. Well known as a redox substrate owing to the redox properties of the nicotinamide ring, β-NAD+ is also fundamental as a substrate of NAD+-consuming enzymes that cleave either high-energy bonds to catalyse their reactions. In this study, a panel of novel adenine-modified NAD+ derivatives was synthesized and biologically evaluated against different NAD+-consuming enzymes. The synthesis of NAD+ derivatives, modified in position 2, 6 or 8 of the adenine ring with aryl/heteroaryl groups, was accomplished by Suzuki-Miyaura cross-couplings. Their biological activity as inhibitors and/or non-natural substrates was assessed against a selected range of NAD+-consuming enzymes. The fluorescence of 8-aryl/heteroaryl NAD+ derivatives allowed their use as biochemical probes for the development of continuous biochemical assays to monitor NAD+-consuming enzyme activities. -
Living with a Cobalamin Cofactor Metabolism Defect Cbl Defects Are
Living with a cobalamin cofactor metabolism defect What you should know about cbl defects that cause homocystinuria The ABCs of cobalamin (cbl) defects Cobalamin cofactor metabolism defects = cbl defects Many different cbl defects cause homocystinuria. (And a few do not.) Each type is named with a letter of the alphabet. Not actual patients or caregivers Marcus has cblC defect Shayna has cblG defect A cbl defect can affect health in many ways Cbl defects impair the body’s ability to metabolize or break down an amino acid called homocysteine. The body makes homocysteine during the metabolism of methionine, another amino acid. Most foods contain methionine. Different forms of cobalamin, also known as vitamin B12, are also involved in the methionine metabolic process. If someone has a combined disorder, then both homocysteine and methylmalonic acid can build up in their blood. If someone has a single disorder, then only homocysteine may build up in their blood. In both types of disorders, serious health problems may develop. Symptoms of combined disorders The most common cbl defect is cblC defect. Like Marcus, most people with cblC defect begin to develop symptoms before they are a year old. This is the early-onset form of cblC defect. As a baby, Marcus was often fussy and didn’t want to eat. Among other symptoms that developed over time, Marcus’s parents began to notice that his eyes wandered. People with late-onset cblC defect may not develop symptoms until later in life – from childhood to adulthood. Late-onset symptoms may be milder than early- BRAIN & SPINAL CORD GROWTH & FEEDING onset symptoms. -

Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— a Literature Review
Molecules 2013, 18, 11873-11903; doi:10.3390/molecules181011873 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— A Literature Review Sowmini Kumaran 1, Mulchand S. Patel 2 and Frank Jordan 1,* 1 Department of Chemistry, Rutgers University, Newark, NJ 07102, USA 2 Department of Biochemistry, School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14214, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-973-353-5470; Fax: +1-973-353-1264. Received: 30 July 2013; in revised form: 14 September 2013 / Accepted: 16 September 2013 / Published: 26 September 2013 Abstract: The 2-oxoacid dehydrogenase complexes (ODHc) consist of multiple copies of three enzyme components: E1, a 2-oxoacid decarboxylase; E2, dihydrolipoyl acyl-transferase; and E3, dihydrolipoyl dehydrogenase, that together catalyze the oxidative decarboxylation of 2-oxoacids, in the presence of thiamin diphosphate (ThDP), coenzyme A 2+ + (CoA), Mg and NAD , to generate CO2, NADH and the corresponding acyl-CoA. The structural scaffold of the complex is provided by E2, with E1 and E3 bound around the periphery. The three principal members of the family are pyruvate dehydrogenase (PDHc), 2-oxoglutarate dehydrogenase (OGDHc) and branched-chain 2-oxo acid dehydrogenase (BCKDHc). In this review, we report application of NMR-based approaches to both mechanistic and structural issues concerning these complexes. These studies revealed the nature and reactivity of transient intermediates on the enzymatic pathway and provided site-specific information on the architecture and binding specificity of the domain interfaces using solubilized truncated domain constructs of the multi-domain E2 component in its interactions with the E1 and E3 components. -

Pyruvate Dehydrogenase Complex Deficiency
Pavlu‑Pereira et al. Orphanet J Rare Dis (2020) 15:298 https://doi.org/10.1186/s13023‑020‑01586‑3 RESEARCH Open Access Pyruvate dehydrogenase complex defciency: updating the clinical, metabolic and mutational landscapes in a cohort of Portuguese patients Hana Pavlu‑Pereira1, Maria João Silva1,2, Cristina Florindo1, Sílvia Sequeira3, Ana Cristina Ferreira3, Sofa Duarte3, Ana Luísa Rodrigues4, Patrícia Janeiro4, Anabela Oliveira5, Daniel Gomes5, Anabela Bandeira6, Esmeralda Martins6, Roseli Gomes7, Sérgia Soares7, Isabel Tavares de Almeida1,2, João B. Vicente8* and Isabel Rivera1,2* Abstract Background: The pyruvate dehydrogenase complex (PDC) catalyzes the irreversible decarboxylation of pyruvate into acetyl‑CoA. PDC defciency can be caused by alterations in any of the genes encoding its several subunits. The resulting phenotype, though very heterogeneous, mainly afects the central nervous system. The aim of this study is to describe and discuss the clinical, biochemical and genotypic information from thirteen PDC defcient patients, thus seeking to establish possible genotype–phenotype correlations. Results: The mutational spectrum showed that seven patients carry mutations in the PDHA1 gene encoding the E1α subunit, fve patients carry mutations in the PDHX gene encoding the E3 binding protein, and the remaining patient carries mutations in the DLD gene encoding the E3 subunit. These data corroborate earlier reports describing PDHA1 mutations as the predominant cause of PDC defciency but also reveal a notable prevalence of PDHX mutations among Portuguese patients, most of them carrying what seems to be a private mutation (p.R284X). The biochemical analyses revealed high lactate and pyruvate plasma levels whereas the lactate/pyruvate ratio was below 16; enzy‑ matic activities, when compared to control values, indicated to be independent from the genotype and ranged from 8.5% to 30%, the latter being considered a cut‑of value for primary PDC defciency.