Aldol Condensation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aldrich FT-IR Collection Edition I Library
Aldrich FT-IR Collection Edition I Library Library Listing – 10,505 spectra This library is the original FT-IR spectral collection from Aldrich. It includes a wide variety of pure chemical compounds found in the Aldrich Handbook of Fine Chemicals. The Aldrich Collection of FT-IR Spectra Edition I library contains spectra of 10,505 pure compounds and is a subset of the Aldrich Collection of FT-IR Spectra Edition II library. All spectra were acquired by Sigma-Aldrich Co. and were processed by Thermo Fisher Scientific. Eight smaller Aldrich Material Specific Sub-Libraries are also available. Aldrich FT-IR Collection Edition I Index Compound Name Index Compound Name 3515 ((1R)-(ENDO,ANTI))-(+)-3- 928 (+)-LIMONENE OXIDE, 97%, BROMOCAMPHOR-8- SULFONIC MIXTURE OF CIS AND TRANS ACID, AMMONIUM SALT 209 (+)-LONGIFOLENE, 98+% 1708 ((1R)-ENDO)-(+)-3- 2283 (+)-MURAMIC ACID HYDRATE, BROMOCAMPHOR, 98% 98% 3516 ((1S)-(ENDO,ANTI))-(-)-3- 2966 (+)-N,N'- BROMOCAMPHOR-8- SULFONIC DIALLYLTARTARDIAMIDE, 99+% ACID, AMMONIUM SALT 2976 (+)-N-ACETYLMURAMIC ACID, 644 ((1S)-ENDO)-(-)-BORNEOL, 99% 97% 9587 (+)-11ALPHA-HYDROXY-17ALPHA- 965 (+)-NOE-LACTOL DIMER, 99+% METHYLTESTOSTERONE 5127 (+)-P-BROMOTETRAMISOLE 9590 (+)-11ALPHA- OXALATE, 99% HYDROXYPROGESTERONE, 95% 661 (+)-P-MENTH-1-EN-9-OL, 97%, 9588 (+)-17-METHYLTESTOSTERONE, MIXTURE OF ISOMERS 99% 730 (+)-PERSEITOL 8681 (+)-2'-DEOXYURIDINE, 99+% 7913 (+)-PILOCARPINE 7591 (+)-2,3-O-ISOPROPYLIDENE-2,3- HYDROCHLORIDE, 99% DIHYDROXY- 1,4- 5844 (+)-RUTIN HYDRATE, 95% BIS(DIPHENYLPHOSPHINO)BUT 9571 (+)-STIGMASTANOL -

Sep. 5, Hydroacylation by Brandon Reinus
Hydroacylation and Related Topics Dong Group Seminar Brandon Reinus Wed, Sept. 5th 2012 Why? ¡ Looking at the reaction, it is a highly atom- economical approach to synthesizing ketones ¡ Umpolung (ex: deprotonating dithioacetals) ¡ Using acrylate derivatives generates a 1,4 diketone relationship, a hard relationship to establish using classical organic synthesis. Presentation Overview 1. Hydroformylation (extremely brief) 2. Rh-Catalyzed Hydroacylation ¡ Intramolecular ¡ Intermolecular ¡ Other 3. NHC Catalyzed Hydroacylation ¡ Benzoin reaction ¡ Stetter reaction ¡ other Part 1 : Background Reppe Roelen Science of Synthesis, Stereoselective Synthesis 1, 2011, pg.409 Hydroformylation Metal-Organic Cooperative Catalysis Park et al. Introduction SCHEME 2 Among the many elegant examples of transition metal cat- alyzed activation of C-HandC-Cbonds,1 chelation-as- sisted protocols have recently attracted increasing attention in organometallic chemistry. Directed metalation processes have been demonstrated by valuable applications in organic synthesis, showing remarkable efficiency and chemoselectivity.2 In general, a chelation-assisted proto- col facilitates the formation of either the kinetically or ther- modynamically favored five- or six-membered metallacycle; aprepositionedcoordinatinggroupinducesspatialproxim- ing decarbonylation, their structures are too specific to apply ity between the C-HorC-Cbondsandthetransitionmetal for common aldehydes. center.1,2 Despite the magnificent usefulness in activating otherwise stable C-HandC-Cbonds,amajordrawbackof -

(Nitroaldol) Reaction
MICROREVIEW DOI: 10.1002/ejoc.201101840 Biocatalytic Approaches to the Henry (Nitroaldol) Reaction Sinéad E. Milner,[a] Thomas S. Moody,[b] and Anita R. Maguire*[c] Keywords: Enzyme catalysis / Biocatalysis / C–C coupling / Nitroaldol reaction / Nitro alcohols Enantiopure β-nitro alcohols are key chiral building blocks approaches to the Henry (nitroaldol) reaction. The first for the synthesis of bioactive pharmaceutical ingredients. method is a direct enzyme-catalysed carbon–carbon bond The preparation of these target compounds in optically pure formation resulting in either an enantio-enriched or enantio- form has been the focus of much research and there has been pure β-nitro alcohol. The second approach describes the an emergence of biocatalytic protocols in the past decade. Henry reaction without stereocontrol followed by a biocata- For the first time, these biotransformations are the focus of lytic resolution to yield the enantiopure β-nitro alcohol. this review. Herein, we describe two principal biocatalytic Introduction The construction of carbon–carbon bonds is an essential element of synthetic organic chemistry. Among the various C–C bond forming reactions, the nitroaldol or Henry reac- tion[1] is one of the classical named reactions in organic synthesis. Essentially, this reaction describes the coupling of a nucleophilic nitro alkane with an electrophilic aldehyde or ketone to produce a synthetically useful β-nitro alcohol (Scheme 1).[2–5] Moreover, the Henry reaction facilitates the joining of two molecular fragments, under mild reaction conditions with the potential formation of two new ste- reogenic centres and a new C–C bond. The resulting β-nitro alcohols can undergo a variety of useful chemical transfor- mations which lead to synthetically useful structural motifs, e.g. -
Stereoselective Reactions of Enolates: Auxiliaries
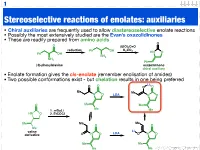
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

Knoevenagel Condensation
Knoevenagel condensation The Knoevenagel condensation (pronounced [ˈknøːvənaːɡl̩ ]) Knoevenagel condensation reaction is an organic reaction named after Emil Knoevenagel. It is a Named after Emil Knoevenagel modification of the aldol condensation.[1][2] Reaction type Coupling reaction A Knoevenagel condensation is a nucleophilic addition of an active Identifiers hydrogen compound to a carbonyl group followed by a dehydration reaction in which a molecule of water is eliminated (hence Organic knoevenagel- condensation). The product is often an α,β-unsaturated ketone (a Chemistry condensation conjugated enone). Portal RSC ontology RXNO:0000044 ID In this reaction the carbonyl group is an aldehyde or a ketone. The catalyst is usually a weakly basic amine. The active hydrogen component has the form[3] Z–CH2-Z or Z–CHR–Z for instance diethyl malonate, Meldrum's acid, ethyl acetoacetate or malonic acid, or cyanoacetic acid.[4] Z–CHR1R2 for instance nitromethane. where Z is an electron withdrawing functional group. Z must be powerful enough to facilitate deprotonation to the enolate ion even with a mild base. Using a strong base in this reaction would induce self-condensation of the aldehyde or ketone. The Hantzsch pyridine synthesis, the Gewald reaction and the Feist–Benary furan synthesis all contain a Knoevenagel reaction step. The reaction also led to the discovery of CS gas. Contents Doebner modification Scope Weiss–Cook reaction See also References Doebner modification The Doebner modification of the Knoevenagel condensation. Acrolein and malonic acid react in pyridine to give trans-2,4-pentadienoic acid with the loss of carbon dioxide. With malonic compounds the reaction product can lose a molecule of carbon dioxide in a subsequent step. -

Knoevenagel Condensation Reaction Catalysed by Agro-Waste Extract As a Greener Solvent Catalyst
ORIGINAL ARTICLE Org. Commun. 14:1 (2021) 81-91 Knoevenagel condensation reaction catalysed by agro-waste extract as a greener solvent catalyst Krishnappa B. Badiger and Kantharaju Kamanna * Peptide and Medicinal Chemistry Research Laboratory, Department of Chemistry, Rani Channamma University, P-B, NH-4, Belagavi 591 156, India (Received January 22, 2021; Revised March 15, 2021; Accepted March 18, 2021) Abstract: This paper present a novel Knoevenagel reaction protocol for the condensation of aromatic/heteroaromatic aldehydes with malononitrile to give α, β–unsaturated benzylidene derivatives. The main focus of this work is to reveal the usability of agro-waste extracts as a catalyst in the Knoevenagel condensation. The present protocol proceeds efficiently for various substituted aromatic and heterocyclic aldehydes in the Knoevenagel reactions. In addition, the present method describes direct isolation of the formed products without using organic solvent extraction gave good yields product. Keywords: one-pot reaction; green chemistry; solvent-free; Knoevenagel condensation reaction; malononitrile; room temperature. ©2021 ACG Publication. All right reserved. 1. Introduction The carbon-carbon bond formation via Knoevenagel condensation1-3 is one of the most important routes repeated in the synthetic organic chemistry and allows the production of various active pharmaceutical molecules.4 Figure 1. Structure of biologically potent benzylidinemalononitrile derivatives * Corresponding author:E-Mail: [email protected] The article was published -

Direct Visualization of a Mechanochemically Induced Molecular Rearrangement Karen J
Angewandte Communications Chemie How to cite: Angew. Chem. Int. Ed. 2020, 59, 13458–13462 Mechanochemical Synthesis International Edition: doi.org/10.1002/anie.201914921 German Edition: doi.org/10.1002/ange.201914921 Direct Visualization of a Mechanochemically Induced Molecular Rearrangement Karen J. Ardila-Fierro, Stipe Lukin, Martin Etter, Krunoslav Uzˇarevic´, Ivan Halasz, Carsten Bolm, and JosØ G. Hernµndez* Abstract: Recent progress in the field of mechanochemistry (thio)acylations[5]). In this context, we became curious as to has expanded the discovery of mechanically induced chemical whether a combination of in situ real-time monitoring by transformations to several areas of science. However, a general synchrotron powder X-ray diffraction (PXRD), Raman fundamental understanding of how mechanochemical reac- spectroscopy, and temperature sensing could be applied for tions by ball milling occur has remained unreached. For this, the investigation of a fundamentally different type of organic we have now implemented in situ monitoring of a mechano- reaction; namely, a molecular rearrangement. As the first chemically induced molecular rearrangement by synchrotron example, we considered the emblematic benzil–benzilic acid X-ray powder diffraction, Raman spectroscopy, and real-time rearrangement in the presence of hydroxide ions (Sche- temperature sensing. The results of this study demonstrate that me 1a). molecular rearrangements can be accomplished in the solid state by ball milling and how in situ monitoring techniques enable the visualization of changes occurring at the exact instant of a molecular migration. The mechanochemical benzil–benzilic acid rearrangement is the focal point of the study. In mechanochemical reactions by ball milling, collisions of accelerated balls inside a milling container maintain a con- stant mixing of the reactants while simultaneously trans- ducing the energy required to induce specific physicochemical changes into the milled system. -

Diversity-Oriented Combinatorial Biosynthesis of Benzenediol Lactone Scaffolds by Subunit Shuffling of Fungal Polyketide Synthases
Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases Yuquan Xua,b,1, Tong Zhouc,1, Shuwei Zhangc, Patricia Espinosa-Artilesb, Luoyi Wangc, Wei Zhanga, Min Lina, A. A. Leslie Gunatilakab,d, Jixun Zhanc,2, and István Molnárb,d,2 aBiotechnology Research Institute, The Chinese Academy of Agricultural Sciences, Beijing 100081, P. R. China; bNatural Products Center, School of Natural Resources and the Environment, University of Arizona, Tucson, AZ 85706; cDepartment of Biological Engineering, Utah State University, Logan, UT 84322; and dBio5 Institute, University of Arizona, Tucson, AZ 85721 Edited by Jerrold Meinwald, Cornell University, Ithaca, NY, and approved June 23, 2014 (received for review April 16, 2014) Combinatorial biosynthesis aspires to exploit the promiscuity of biosynthesis inhibitory activities in animals. 10,11-dehydrocurvularin microbial anabolic pathways to engineer the synthesis of new (7;Fig.1)isaDALwitha12-memberedring(DAL12)that chemical entities. Fungal benzenediol lactone (BDL) polyketides modulates the heat shock response and the immune system (8, 9). are important pharmacophores with wide-ranging bioactivities, BDL scaffolds are biosynthesized by pairs of collaborating, including heat shock response and immune system modulatory sequentially acting iterative polyketide synthases (iPKSs) (3) – effects. Their biosynthesis on a pair of sequentially acting iterative forming quasi-modular BDL synthases (BDLSs) (Fig. 1) (11 14). polyketide synthases (iPKSs) offers a test case for the modulariza- Each of the BDLS subunits catalyze recursive, decarboxylative tion of secondary metabolic pathways into “build–couple–pair” Claisen condensations of malonyl-CoA using a single core set of ketoacyl synthase (KS), acyl transferase (AT), and acyl carrier combinatorial synthetic schemes. -

Experiment 19 — Aldol Condensation
Chem 22 Spring 2010 Experiment 19 — Aldol Condensation _____________________________________________________________________________ Pre-lab preparation. (1) Write the mechanism of the base-catalyzed aldol condensation of acetone and a generalized aromatic aldehyde, Ar–CH=O to give the α,β-unsaturated product (i.e. the reaction at the top of the next page, but with a 1:1 ratio of ketone and aldehyde). Remember that dehydration in this case occurs under basic conditions, so it can't start with protonation of the hydroxyl group. Nor can it go via an E2 pathway. (2) Draw the structures of all the possible aldehyde and ketone reactants (not the 25 possible condensation products!). (3) The new CC double bonds of the condensation products are E rather than Z, as shown in the acetone example. Why? (4) What's the purpose of rinsing the crude product with dilute acetic acid, followed by ethanol? (5) What is the procedure for carrying out a single-solvent recrystallization? Write this out in detail. The aldol condensation has historically been one of the favorite tools in the synthetic organic chemist's repertoire because of its versatility in forming new CC bonds. Since its discovery in the 1870s the aldol condensation has been use extensively in the synthesis of natural products and other complex molecules. In a typical base-catalyzed aldol condensation an enolate ion attacks the carbonyl group of an aldehyde or ketone. This carbonyl addition produces a β-hydroxy carbonyl compound. In many cases the initially formed condensation product undergoes an "E1cB" dehydration to produce an α,β-unsaturated carbonyl compound as the final product. -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities.