Strategies Towards Asymmetric Conjugate Trifluoromethylation of Α
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UMPOLUNG in REACTIONS CATALYZED by THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung En Reacciones Catalizadas Por Enzimas Dependientes De Pirofosfato De Tiamina
Ciencia, Ambiente y Clima, Vol. 2, No. 2, julio-diciembre, 2019 • ISSN (impreso): 2636-2317 • ISSN (en línea): 2636-2333 DOI: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 UMPOLUNG IN REACTIONS CATALYZED BY THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung en reacciones catalizadas por enzimas dependientes de pirofosfato de tiamina Carlos José Boluda Emily Soto Instituto Tecnológico de Santo Domingo (INTEC), Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales, Av. de Los Área de Ciencias Básicas y Ambientales Próceres 49, Santo Domingo, República Dominicana Correo-e: [email protected] *Corresponding author: Carlos J. Boluda Darah de la Cruz Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Carolina Juncá Área de Ciencias Básicas y Ambientales Instituto Tecnológico de Santo Domingo (INTEC), Correo-e: [email protected] Área de Ciencias Básicas y Ambientales Anny Peña Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales Correo-e: [email protected] Recibido: 25/9/2019 • Aprobado: 19/10/2019 Cómo citar: Boluda, C. J., Juncá, C., Soto, E., de la Cruz, D., & Peña, A. (2019). Umpolung in reactions catalyzed by thiamine pyrophos- phate dependent enzymes. Ciencia, Ambiente Y Clima, 2(2), 27-42. Doi: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 Abstract Resumen The temporal exchange of the electrophilic/nucleophilic El intercambio temporal del carácter electrofílico/nucleofí- character of an atom by chemical manipulation is known lico de un átomo mediante manipulación química, es cono- in organic chemistry as umpolung. This inversion of polarity cido con el vocablo alemán de umpolung. -
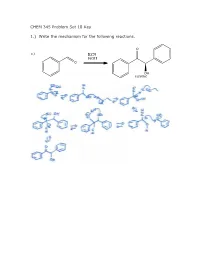
CHEM 345 Problem Set 18 Key 1.) Write the Mechanism for The
CHEM 345 Problem Set 18 Key 1.) Write the mechanism for the following reactions. O a.) KCN EtOH O OH racemic 1.) Write the mechanism for the following reactions. b.) KCN AcOH O NC OH racemic O c.) N S R O NEt3 OH racemic 2.) What is the structure of AcOH?Why does changing the solvent from EtOH to AcOH make such a big difference? O OH AcOH acetic acid The pKa of acetic acid is approximately 5. The pKa of ethanol is approximately 15. When you take a proton off of ethanol, you generate ethoxide which is about 1010 times stronger of a base than acetate. 3.) Give two instances when you need to use the thiazolium salt and triethylamine rather than KCN and EtOH. If the aldehydes contain an enolizable proton then you cannot use KCN/EtOH, instead you must use the thiazolium. Also, if the electrophile is a Michael acceptor to give a 1,4 dicarbonyl, then the thiazolium catalyst should be used. 4.) Break the following compound down as far as you can using Aldol, Michael, and Claisen reactions. Above each retrosynthetic arrow, write the name of the reaction. O HO O HO Aldol O Michael HO O O HO O Aldol O O Aldol HO O O HO Michael O O O Aldol There are other possibilities for order. O O HO Aldol O O 5.) Synthesize the following molecules. All carbons in the molecules must come from benzene or compounds with 5C’s or less. a.) O H2SO4 O HNO3 O2N AlCl3 O O SOCl2 HO Cl H2CrO4 1.) BuLi, Et2O + O 2.) H3O HO b.) O O O Cl + H3O NaOEt, EtOH O O O O Cl O AlCl3 Cl Cl AlCl3 Cl2 c.) O OMe NaOMe MeOH O O 1.) POCl3, DMF 2.) H2O OMe OMe O AlCl3 MeI Cl ONa HCl NaOH ZnHg 1.) NaOH + mcpba O 2.) H3O O OH O O AlCl3 Cl 6.) Write the mechanism for the following reactions. -

Shelf-Stable Electrophilic Trifluoromethylating Reagents: a Brief Historical Perspective
Shelf-stable electrophilic trifluoromethylating reagents: A brief historical perspective Norio Shibata*1, Andrej Matsnev1 and Dominique Cahard*2 Review Open Access Address: Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65 1Department of Frontier Materials, Graduate School of Engineering, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya Received: 15 March 2010 466-8555, Japan and 2UMR 6014 CNRS – C.O.B.R.A. Université et Accepted: 21 May 2010 INSA de Rouen, 1 rue Tesnière, 76130 Mont Saint Aignan, France Published: 16 June 2010 Email: Guest Editor: D. O'Hagan Norio Shibata* - [email protected]; Dominique Cahard* - [email protected] © 2010 Shibata et al; licensee Beilstein-Institut. License and terms: see end of document. * Corresponding author Keywords: asymmetric synthesis; electrophilic; fluorine; reagent; trifluoromethylation Abstract Since the discovery by Yagupolskii and co-workers that S-trifluoromethyl diarylsulfonium salts are effective for the trifluoro- methylation of thiophenolates, the design and synthesis of electrophilic trifluoromethylating reagents have been extensively researched in both academia and industry, due to the significant unique features that trifluoromethylated compounds have in phar- maceuticals, agricultural chemicals, and functional materials. Several effective reagents have been developed by the groups of Yagupolskii, Umemoto, Shreeve, Adachi, Magnier, Togni and Shibata. Due to the high stability and reactivity of these reagents, a series of Umemoto reagents, Togni reagent and Shibata reagent are now commercially available. In this review, we wish to briefly provide a historical perspective of the development of so-called “shelf-stable electrophilic trifluoromethylating reagents”, although this field is in constant development. Review The chemistry of fluoro-organic compounds is one of the areas [3]. -

Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— a Literature Review
Molecules 2013, 18, 11873-11903; doi:10.3390/molecules181011873 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— A Literature Review Sowmini Kumaran 1, Mulchand S. Patel 2 and Frank Jordan 1,* 1 Department of Chemistry, Rutgers University, Newark, NJ 07102, USA 2 Department of Biochemistry, School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14214, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-973-353-5470; Fax: +1-973-353-1264. Received: 30 July 2013; in revised form: 14 September 2013 / Accepted: 16 September 2013 / Published: 26 September 2013 Abstract: The 2-oxoacid dehydrogenase complexes (ODHc) consist of multiple copies of three enzyme components: E1, a 2-oxoacid decarboxylase; E2, dihydrolipoyl acyl-transferase; and E3, dihydrolipoyl dehydrogenase, that together catalyze the oxidative decarboxylation of 2-oxoacids, in the presence of thiamin diphosphate (ThDP), coenzyme A 2+ + (CoA), Mg and NAD , to generate CO2, NADH and the corresponding acyl-CoA. The structural scaffold of the complex is provided by E2, with E1 and E3 bound around the periphery. The three principal members of the family are pyruvate dehydrogenase (PDHc), 2-oxoglutarate dehydrogenase (OGDHc) and branched-chain 2-oxo acid dehydrogenase (BCKDHc). In this review, we report application of NMR-based approaches to both mechanistic and structural issues concerning these complexes. These studies revealed the nature and reactivity of transient intermediates on the enzymatic pathway and provided site-specific information on the architecture and binding specificity of the domain interfaces using solubilized truncated domain constructs of the multi-domain E2 component in its interactions with the E1 and E3 components. -

Perfluoro-3-Ethyl-2,4-Dimethyl-3
Journal of Fluorine Chemistry 227 (2019) 109370 Contents lists available at ScienceDirect Journal of Fluorine Chemistry journal homepage: www.elsevier.com/locate/fluor Perfluoro-3-ethyl-2,4-dimethyl-3-pentyl persistent radical: A new reagent T for direct, metal-free radical trifluoromethylation and polymer initiation ⁎ ⁎ ⁎ Haibo Meia, Jianlin Hana, , Sarah Whiteb, , Greg Butlerb, Vadim A. Soloshonokc,d, a College of Chemical Engineering, Nanjing Forestry University, Nanjing, 210037, China b Oakwood Chemical, Inc. 730 Columbia Hwy. N, Estill, SC, 29918, USA c Department of Organic Chemistry I, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel Lardizábal 3, 20018, San Sebastián, Spain d IKERBASQUE, Basque Foundation for Science, María Díaz de Haro 3, Plaza Bizkaia, 48013, Bilbao, Spain ARTICLE INFO ABSTRACT Keywords: This review comprehensively profiles perfluoro-3-ethyl-2,4-dimethyl-3-pentyl persistent radical (PPFR) asanew Fluorine reagent for radical trifluoromethylation, trifluoromethylation/fluorination and polymer initiation. The PPFRis Trifluoromethylation perfectly stable at ambient conditions, but at temperatures above 80 °C undergoes β-scission to generate tri- Persistent radical fluoromethyl radical. This property can be used to initiate various chain-polymerization or trifluoromethylation Fluorination reactions. The unique feature of this process, distinguishing it from all other known methods for ·CF3-radical Polymer generation, is that the radical is produced under neutral, inert and additive-free -

Advances in Catalytic Enantioselective Fluorination, Mono‑,Di‑, and Trifluoromethylation, and Trifluoromethylthiolation Reactions ‡ ‡ † Xiaoyu Yang, Tao Wu, Robert J
Review pubs.acs.org/CR Advances in Catalytic Enantioselective Fluorination, Mono‑,Di‑, and Trifluoromethylation, and Trifluoromethylthiolation Reactions ‡ ‡ † Xiaoyu Yang, Tao Wu, Robert J. Phipps, and F. Dean Toste* Department of Chemistry, University of California, Berkeley, California 94720, United States 6. Catalytic Enantioselective Trifluoromethylthiola- tion 865 7. Summary and Outlook 866 Author Information 866 Corresponding Author 866 Present Address 866 Author Contributions 866 Notes 866 CONTENTS Biographies 866 Acknowledgments 867 1. Introduction 826 References 867 2. Catalytic Enantioselective Fluorination 827 2.1. Electrophilic Fluorination 827 2.1.1. Metal-Catalyzed Fluorination Involving 1. INTRODUCTION Enolates 827 Despite being largely absent from natural products and 2.1.2. Metal-Catalyzed Fluorination Not In- biological processes, fluorine plays a conspicuous and volving Enolates 834 increasingly important role within pharmaceuticals and agro- 2.1.3. Organocatalytic Electrophilic Fluorina- − chemicals, as well as in materials science.1a c Indeed, as many as tion 835 35% of agrochemicals and 20% of pharmaceuticals on the 2.1.4. Fluorination Using Multiple Catalysts 846 market contain fluorine.1d Fluorine is the most electronegative 2.1.5. One-Pot and Tandem Processes 848 element in the periodic table, and the introduction of one or 2.2. Nucleophilic Fluorination 850 more fluorine atoms into a molecule can result in greatly 3. Catalytic Enantioselective Trifluoromethylation perturbed properties. Fluorine substituents can potentially and Perfluoroalkylation 853 impact a number of variables, such as the acidity or basicity of 3.1. Asymmetric Nucleophilic Trifluoromethyla- neighboring groups, dipole moment, and properties such as tion 853 lipophilicity, metabolic stability, and bioavailability. The 3.1.1. Overview of Nucleophilic Trifluorome- multitude of effects that can arise from the introduction of thylation 853 fluorine in small molecules in the context of medicinal 3.1.2. -

CV-PSB-August 2020
Curriculum Vitae Phil S. Baran Appointment: Scripps Research Professor, Department of Chemistry 10550 North Torrey Pines Road, BCC-436 La Jolla, California 92037 Telephone: (858) 784-7373 Facsimile: (858) 784-7575 Email: [email protected] Website: www.scripps.edu/chem/baran/ January, 2013 Darlene Shiley Professor of Chemistry April, 2009 Member, Skaggs Institute for Chemical Biology June, 2008 Professor of Chemistry July, 2006 Associate Professor of Chemistry (with Tenure) June, 2003 Assistant Professor of Chemistry Date/Place of Birth: 10 Aug 1977 / Denville, NJ, USA Citizenship: United States Education 2001 – 2003 Postdoctoral Associate Advisor: Professor E.J. Corey Harvard University, Cambridge, Massachusetts 1997 – 2001 Ph.D. Graduate Student in Chemistry Advisor: Professor K.C. Nicolaou The Scripps Research Institute, La Jolla, California 1995 – 1997 B.S. with Honors in Chemistry Advisor: Professor D.I. Schuster New York University, New York, New York 1991 – 1995 Simultaneous high school graduation from Mt. Dora High School and A.A. degree with honors, Lake Sumter Community College, Florida Awards • Inhoffen Medal, 2019 • Manchot Research Professorship, 2017 • Member, The National Academy of Sciences, 2017 • Emanuel Merck Lectureship, 2017 • Blavatnik National Laureate in Chemistry, 2016 • ACS Elias J. Corey Award, 2016 • Member, American Academy of Arts and Sciences, 2015 • College of Arts and Science Alumni Distinguished Service Award, New York University, 2015 • Reagent of the Year Award (EROS), 2015 • Mukaiyama Award, 2014 • -

Development of Palladium-Catalyzed Decarboxylative Allylation of Electron-Deficient Sulfones: Method Development and Mechanistic Studies
Development of Palladium-Catalyzed Decarboxylative Allylation of Electron-Deficient Sulfones: Method Development and Mechanistic Studies by Monica Anne Gill A thesis submitted to the Faculty of Graduate and Postdoctoral Affairs in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry Carleton University Ottawa, Ontario © 2015, Monica Anne Gill Abstract Palladium-catalyzed decarboxylative allylation is a powerful method of carbon-carbon bond construction. This methodology relies on an electron- withdrawing group to promote the reaction. The use of sulfones in decarboxylative allylation has been explored using the trifluoromethylsulfonyl (triflyl) group, as well as the bis(3,5-trifluoromethyl)phenylsulfonyl (BTMP) group. These substrates are highly reactive at room temperature (triflyl) and 50 oC (BTMP sulfones). A detailed mechanistic study using deuterium-labelled substrates was performed to understand the origin of the protonation side-product. It was proposed that a β- hydride elimination from the η1 allyl on palladium could generate a palladium hydride intermediate along with an allene. Although small amounts of deuterium incorporation were observed in the protonated products, the proposed mechanism could not be the major pathway. Using isotopically labelled ligand, however, all protonation was supressed. This suggests that the origin of the proton is actually from the ligand and that kinetic isotope effects may be responsible for inhibiting the protonation pathway with labelled ligand. ii Acknowledgments I would like to thank Dr. Jeff Manthorpe for giving me the chance to pursue my PhD in his lab. I always appreciated Jeff’s enthusiasm for organic chemistry as well as his vast knowledge of the subject. -

Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids
International Journal of Molecular Sciences Article Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids Stefan R. Marsden, Duncan G. G. McMillan and Ulf Hanefeld * Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Van der Maasweg 9, 2629HZ Delft, The Netherlands; [email protected] (S.R.M.); [email protected] (D.G.G.M.) * Correspondence: [email protected] Received: 12 October 2020; Accepted: 12 November 2020; Published: 16 November 2020 Abstract: The synthetic properties of the Thiamine diphosphate (ThDP)-dependent pyruvate dehydrogenase E1 subunit from Escherichia coli (EcPDH E1) was assessed for carboligation reactions with aliphatic ketoacids. Due to its role in metabolism, EcPDH E1 was previously characterised with respect to its biochemical properties, but it was never applied for synthetic purposes. Here, we show that EcPDH E1 is a promising biocatalyst for the production of chiral α-hydroxyketones. WT EcPDH E1 shows a 180–250-fold higher catalytic efficiency towards 2-oxobutyrate or pyruvate, respectively, in comparison to engineered transketolase variants from Geobacillus stearothermophilus (TKGST). Its broad active site cleft allows for the efficient conversion of both (R)- and (S)-configured α-hydroxyaldehydes, next to linear and branched aliphatic aldehydes as acceptor substrates under kinetically controlled conditions. The alternate, thermodynamically controlled self-reaction of aliphatic aldehydes was shown to be limited to low levels of conversion, which we propose to be due to their large hydration constants. Additionally, the thermodynamically controlled approach was demonstrated to suffer from a loss of stereoselectivity, which makes it unfeasible for aliphatic substrates. Keywords: Thiamine diphosphate; transketolase; C-C bond formation; kinetic control; acyloins 1. -
![Umpolung of Amine Reactivity. Nucleophilic [Alpha]](https://docslib.b-cdn.net/cover/9062/umpolung-of-amine-reactivity-nucleophilic-alpha-2689062.webp)
Umpolung of Amine Reactivity. Nucleophilic [Alpha]
11591 S. W. Benson: Thermochemical Kinetics. Wiley, New York, N. Y., [I631 C. M. Shy and J. F. Finklea, Environ. Sci. Technol. 7, 205 (1973). 1968. [I641 R. P. Steer, K. R. Darnall, and J. N. Pifrs, Jr., Tetrahedron Lett. [I601 J. G. Caluert, K. L. Demerjian, and J. A. Kerr, Proc. Int. Symp. 1969. 3765. Air Pollut., Tokyo, Oct. 17-19, 1972, pp. 465ff. 11651 S. Furuyama. R. Atkinson, A. J. Colussi, and R. J. Cvetanouic, Int. [161] A. Q. Eschenroeder and J. R. Marfinez, Advan. Chem. Ser. 113, 101 J. Chem. Kinet. 6, 741 (1974). (1972). [166] J. N. Pitrs, Jr., P. G. Bekowies. G. J. Doyle, J. M. McAfee, and [I621 7: A. Hecht, J. H. Seinfeld. and M. C. Dodge, Environ. Sci. Technol. A. M. Winer, to be oublished. 8, 327 (1974). and references therein. Umpolung of Amine Reactivity. Nucleophilic a-(Secondary Amino)- New synthetic alky lation via Metalated Nitrosaminesr**lr”’l methods 0 By Dieter Seebach and Dieter Ended*] There are basically two kinds of hetero atoms in organic molecules: one kind confers electrophilic character upon the carbon atom to which it is bound, and the other kind turns it into a nucleophilic site. The development of methods permitting transitions between the two resulting categories of reagents has become an important task of modern organic synthesis. The scope of such umpolung of the reactivity of functional groups is discussed for the case of amines as an example. A method of preparing masked u-secondary amino carbanions consists in nitrosation of the secondary amine, followed by metalation of the resulting nitrosamine CL to the nitrogen, reaction with electrophiles, and subsequent denitrosation. -

Organic Chemistry 2 Ester Enolates and Acyl Anion Equivalents
Dr. Peter Wipf Chemistry 0320 - Organic Chemistry 2 Ester Enolates and Acyl Anion Equivalents The Claisen condensation is the ester analog of the aldol reaction. Under standard conditions (sodium alkoxide, alkohol), the equilibrium is shifted to the β-dicarbonyl compound as a consequence of the irreversible enolization to the highly resonance-stabilized dicarbonyl enolate. O O NaOR, HOR O O O O + OR R' R' R' R'=OR, C The intramolecular Claisen reaction is generally called the Dieckmann condensation. Another modification of the Claisen condensation is the use of strong base which leads to irreversible enolization of the carbonyl compounds. O OLi LDA, -78 °C PhCOCl O O OEt OEt THF Ph OEt β-Dicarbonyl, and, generally, active methylene compounds provide a rich source of chemistry in alkylation reactions, due to their relative ease of enolization and the possibility for removal of one of the activating groups. Z1 Z2 , Z = ester, ketone, amide, nitrile, aldehyde, nitro, sulfoxide, sulfone, sulfonate, sulfonamide H H Especially important are acetoacetate and malonate building blocks (acetoacetic and malonic ester syntheses): O O O O O O OEt OEt OEt XCH2CO2R R-X R-X (1 equiv) RCOX O O O O OH O O R R OEt O R Dithioketals can be used in Umpolung reactions as latent carbonyl groups. Nature’s Umpolung reagent is thiamine pyrophosphate (TPP). In the pentose phosphate pathway, TPP catalyzes the conversion of erythrose into fructose. First, the carbanion of TPP attacks the carbonyl carbon of xylulose 5-phosphate, forming a proton from the TPP adduct, and subsequent electron rearrangement leads to fragmentation, releasing the first product, glyceraldehyde 3-phosphate. -
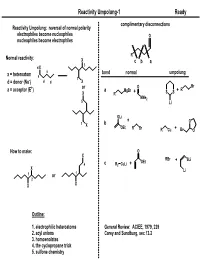
Reactivity Umpolung-1
Reactivity Umpolung-1 Ready complimentary disconnections Reactivity Umpolung: reversal of normal polarity electrophiles become nucleophiles O nucleophiles become electrophiles R Normal reactivity: X c ba 3 d X a bond normal umpolung x = heteroatom a 1 d = donor (Nu-) d d X + or O Br a = acceptor (E ) a MgBr + R X R + SS NMe2 5 Li OLi + O 1 b X OEt R Br R Cu + Br O How to make: O X RBr OEt + SLi 4 c R2 CuLi + X Li 1 or 1 2 X X Outline: 1. electrophilic heteroatoms General Review: ACIEE, 1979, 239 2. acyl anions Carey and Sundburg, sec 13.2 3. homoenolates 4. the cyclopropane trick 5. sulfone chemistry Reactivity Umpolung-2 Ready Electrophilic Heteroatoms mCPBA 'Rubottom [O]' OTMS 1. mCPBA OTMS O General form: 2. HF Ph Ph Ph 60% O OH X X Y+ X,Y = heteroatoms Y Rubottom, TL, 1974, 4319 Electrophile Examples O O O O2 O O 1. LDA; O2 O 1. TBSOTf, Et3N DMDO PO PO OTBS NMe2 2. Na2S2O3 HO 2. DMDO NMe2 84% P'O OP" 69% P'O OP" OP" O OP" HO O NMe2 Falck, JOC, 1995, 3385 Oxaziridines 1. LiHMDS 2. " HO Ph CO2Et Ph CO2Et 81% O O N OH O S O Wasserman and Lipshutz O TL, 1975, 1731 O 95% ee O O Mo O O Davis, Chem. Rev. 1992, 919 O O OH Py O LDA; MoOPH not a general asymmetric method. For racemic, most 87% commonly used is 'Davis Oxaziridine' P(NMe2)3 O MoOPH Ph Vedejs, JOC, 1978, 188 TsN Reactivity Umpolung-3 Ready Aminations (Review: Syn Lett, 1997, 741) Heteroatom exchange: Most commonly with Br, Cl Electrophile Examples O O cat PCl3 O NH3 R R R OH E OH OH Br2 OH N 1.