Chem. Sci., 3, 633-658
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Shelf-Stable Electrophilic Trifluoromethylating Reagents: a Brief Historical Perspective
Shelf-stable electrophilic trifluoromethylating reagents: A brief historical perspective Norio Shibata*1, Andrej Matsnev1 and Dominique Cahard*2 Review Open Access Address: Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65 1Department of Frontier Materials, Graduate School of Engineering, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya Received: 15 March 2010 466-8555, Japan and 2UMR 6014 CNRS – C.O.B.R.A. Université et Accepted: 21 May 2010 INSA de Rouen, 1 rue Tesnière, 76130 Mont Saint Aignan, France Published: 16 June 2010 Email: Guest Editor: D. O'Hagan Norio Shibata* - [email protected]; Dominique Cahard* - [email protected] © 2010 Shibata et al; licensee Beilstein-Institut. License and terms: see end of document. * Corresponding author Keywords: asymmetric synthesis; electrophilic; fluorine; reagent; trifluoromethylation Abstract Since the discovery by Yagupolskii and co-workers that S-trifluoromethyl diarylsulfonium salts are effective for the trifluoro- methylation of thiophenolates, the design and synthesis of electrophilic trifluoromethylating reagents have been extensively researched in both academia and industry, due to the significant unique features that trifluoromethylated compounds have in phar- maceuticals, agricultural chemicals, and functional materials. Several effective reagents have been developed by the groups of Yagupolskii, Umemoto, Shreeve, Adachi, Magnier, Togni and Shibata. Due to the high stability and reactivity of these reagents, a series of Umemoto reagents, Togni reagent and Shibata reagent are now commercially available. In this review, we wish to briefly provide a historical perspective of the development of so-called “shelf-stable electrophilic trifluoromethylating reagents”, although this field is in constant development. Review The chemistry of fluoro-organic compounds is one of the areas [3]. -

Recent Advances on Mechanistic Studies on C–H Activation
Open Chem., 2018; 16: 1001–1058 Review Article Open Access Daniel Gallego*, Edwin A. Baquero Recent Advances on Mechanistic Studies on C–H Activation Catalyzed by Base Metals https:// doi.org/10.1515/chem-2018-0102 received March 26, 2018; accepted June 3, 2018. 1Introduction Abstract: During the last ten years, base metals have Application in organic synthesis of transition metal- become very attractive to the organometallic and catalytic catalyzed cross coupling reactions has been positioned community on activation of C-H bonds for their catalytic as one of the most important breakthroughs during the functionalization. In contrast to the statement that new millennia. The seminal works based on Pd–catalysts base metals differ on their mode of action most of the in the 70’s by Heck, Noyori and Suzuki set a new frontier manuscripts mistakenly rely on well-studied mechanisms between homogeneous catalysis and synthetic organic for precious metals while proposing plausible chemistry [1-5]. Late transition metals, mostly the precious mechanisms. Consequently, few literature examples metals, stand as the most versatile catalytic systems for a are found where a thorough mechanistic investigation variety of functionalization reactions demonstrating their have been conducted with strong support either by robustness in several applications in organic synthesis [6- theoretical calculations or experimentation. Therefore, 12]. Owing to the common interest in the catalysts mode we consider of highly scientific interest reviewing the of action by many research groups, nowadays we have a last advances on mechanistic studies on Fe, Co and Mn wide understanding of the mechanistic aspects of precious on C-H functionalization in order to get a deep insight on metal-catalyzed reactions. -

Perfluoro-3-Ethyl-2,4-Dimethyl-3
Journal of Fluorine Chemistry 227 (2019) 109370 Contents lists available at ScienceDirect Journal of Fluorine Chemistry journal homepage: www.elsevier.com/locate/fluor Perfluoro-3-ethyl-2,4-dimethyl-3-pentyl persistent radical: A new reagent T for direct, metal-free radical trifluoromethylation and polymer initiation ⁎ ⁎ ⁎ Haibo Meia, Jianlin Hana, , Sarah Whiteb, , Greg Butlerb, Vadim A. Soloshonokc,d, a College of Chemical Engineering, Nanjing Forestry University, Nanjing, 210037, China b Oakwood Chemical, Inc. 730 Columbia Hwy. N, Estill, SC, 29918, USA c Department of Organic Chemistry I, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel Lardizábal 3, 20018, San Sebastián, Spain d IKERBASQUE, Basque Foundation for Science, María Díaz de Haro 3, Plaza Bizkaia, 48013, Bilbao, Spain ARTICLE INFO ABSTRACT Keywords: This review comprehensively profiles perfluoro-3-ethyl-2,4-dimethyl-3-pentyl persistent radical (PPFR) asanew Fluorine reagent for radical trifluoromethylation, trifluoromethylation/fluorination and polymer initiation. The PPFRis Trifluoromethylation perfectly stable at ambient conditions, but at temperatures above 80 °C undergoes β-scission to generate tri- Persistent radical fluoromethyl radical. This property can be used to initiate various chain-polymerization or trifluoromethylation Fluorination reactions. The unique feature of this process, distinguishing it from all other known methods for ·CF3-radical Polymer generation, is that the radical is produced under neutral, inert and additive-free -

2.1 the 18-ELECTRON RULE the 18E Rule1 Is a Way to Help Us Decide
30 GENERAL PROPERTIES OF ORGANOMETALLIC COMPLEXES 2.1 THE 18-ELECTRON RULE The 18e rule1 is a way to help us decide whether a given d-block transition metal organometallic complex is likely to be stable. Not all the organic formulas we can write down correspond to stable species. For example, CH5 requires a 5-valent carbon and is therefore not stable. Stable compounds, such as CH4,have the noble gas octet, and so carbon can be thought of as following an 8e rule. This corresponds to carbon using its s and three p orbitals to form four filled bonding orbitals and four unfilled antibonding orbitals. On the covalent model, we can consider that of the eight electrons required to fill the bonding orbitals, four come from carbon and one each comes from the four H substituents. We can therefore think of each H atom as being a 1e ligand to carbon. To assign a formal oxidation state to carbon in an organic molecule, we impose an ionic model by artificially dissecting it into ions. Each electron pair in any bond is assigned to the most electronegative of the two atoms or groups that constitute the bond. For methane, this dissection gives C4− + 4H+, with carbon as the more electronegative element. This makes methane an 8e compound with an oxidation state of −4, usually written C(-IV). Note that the net electron count always remains the same, whether we adopt the covalent (4e {Catom}+4 × 1e {4H atoms}=8e) or ionic (8e{C4−ion}+4 × 0e{4H+ions}=8e) model. -

Assessing the Feasibility of a One-Pot, Tandem Olefin Metathesis and Isomerization Sequence to Synthesize Conjugated Aromatic Olefins
University of Tennessee at Chattanooga UTC Scholar Student Research, Creative Works, and Honors Theses Publications 8-2016 Assessing the feasibility of a one-pot, tandem olefin metathesis and isomerization sequence to synthesize conjugated aromatic olefins Ajay D. Makwana University of Tennessee at Chattanooga, [email protected] Follow this and additional works at: https://scholar.utc.edu/honors-theses Part of the Chemistry Commons Recommended Citation Makwana, Ajay D., "Assessing the feasibility of a one-pot, tandem olefin metathesis and isomerization sequence to synthesize conjugated aromatic olefins" (2016). Honors Theses. This Theses is brought to you for free and open access by the Student Research, Creative Works, and Publications at UTC Scholar. It has been accepted for inclusion in Honors Theses by an authorized administrator of UTC Scholar. For more information, please contact [email protected]. Assessing the Feasibility of a One-Pot, Tandem Olefin Metathesis and Isomerization Sequence to Synthesize Conjugated Aromatic Olefins Ajay D. Makwana Departmental Honors Thesis The University of Tennessee at Chattanooga Department of Chemistry Project Director: Dr. Kyle S. Knight Examination Date: April 4, 2016 Members of Examination Committee: Dr. John P. Lee Dr. Han J. Park Dr. David Giles 1 Abstract The synthesis of substituted phenylpropene dimers using a one-pot, tandem olefin metathesis and isomerization sequence has been studied. This sequence relies on the facilitated, in-situ conversion of a ruthenium carbene species (Ru=C) to a ruthenium hydride species (Ru-H) upon addition of an inorganic hydride source. Three separate reactions occur within one reaction flask: 1) olefin metathesis of the starting phenylpropene to yield phenylpropene dimer via Ru=C catalyst, 2) conversion of Ru=C to Ru-H via addition of an inorganic hydride source, 3) isomerization of phenylpropene dimer via insertion and β-hydride elimination to yield conjugated product. -

Wilkinson's Catalyst
Homogeneous Catalytic Processes Hydroformylation RCH2CH2 O RCH CH2 + CO + H2 Co(I), Rh(I) or Pt(II) H Oxidation H3C O H2CCH2 + O2 Pd(II) or Cu(II) H Carbonylation H3C O - CH3OH + CO [RhI2(CO)2] OH Hydrocyanation [Ni{P(OR) } ] H CCC CH + 2HCN 3 4 NCCH CH CH CH CN 2 H H 2 2 2 2 2 Cyclotrimerization Ni(acac) 3 HC CH 2 Hydrogenation of Alkenes The most commonly used catalyst is the Wilkinson’s Catalyst Many alkenes are hydrogenated with hydrogen at 1atm pressure or less . Wilkinson’s catalyst is highly sensitive to the nature of the phosphine ligand and the alkene substrate. Analogous catalysts with alkyl phosphine ligands are inactive Highly hindered alkenes and ethylene are not hydrogenated by the catalyst Wilkinson’s Catalyst Ph3P PPh3 R CH3 Rh H2 Ph3P Cl Reductive Elimination Oxidative Addition R H H PPh3 H PPh3 Rh Rh Ph3P Cl Ph3P Cl PPh3 PPh3 Ligand Dissociation Ligand Association PPh3 PPh3 H R H PPh3 Rh H PPh3 Cl Rh Ph3P Cl PPh3 H Migratory Insertion H PPh3 Rh R Ph3P Cl Alkene Coordination R Hydroformylation Catalyst + CO + H2 RCH2CH2CHO R A less common,,pppyy but more appropriate name is hydrocarbonylation Both cobalt and rhodium complexes are used as catalysts. Alkene iitiisomerization, alkene hdhydrogena tion and ftiformation of bhdbranched alde hy des are the possible side reactions. Cobalt catalysts operate at 150 ºC and 250 atm, whereas Rhodium catalysts operate at moderate temperatures and 1 atm. Rhodium catalysts promotes the formation of linear aldehydes. Cobalt catalysts do so if modified with alkylphosphine ligands. -

Advances in Catalytic Enantioselective Fluorination, Mono‑,Di‑, and Trifluoromethylation, and Trifluoromethylthiolation Reactions ‡ ‡ † Xiaoyu Yang, Tao Wu, Robert J
Review pubs.acs.org/CR Advances in Catalytic Enantioselective Fluorination, Mono‑,Di‑, and Trifluoromethylation, and Trifluoromethylthiolation Reactions ‡ ‡ † Xiaoyu Yang, Tao Wu, Robert J. Phipps, and F. Dean Toste* Department of Chemistry, University of California, Berkeley, California 94720, United States 6. Catalytic Enantioselective Trifluoromethylthiola- tion 865 7. Summary and Outlook 866 Author Information 866 Corresponding Author 866 Present Address 866 Author Contributions 866 Notes 866 CONTENTS Biographies 866 Acknowledgments 867 1. Introduction 826 References 867 2. Catalytic Enantioselective Fluorination 827 2.1. Electrophilic Fluorination 827 2.1.1. Metal-Catalyzed Fluorination Involving 1. INTRODUCTION Enolates 827 Despite being largely absent from natural products and 2.1.2. Metal-Catalyzed Fluorination Not In- biological processes, fluorine plays a conspicuous and volving Enolates 834 increasingly important role within pharmaceuticals and agro- 2.1.3. Organocatalytic Electrophilic Fluorina- − chemicals, as well as in materials science.1a c Indeed, as many as tion 835 35% of agrochemicals and 20% of pharmaceuticals on the 2.1.4. Fluorination Using Multiple Catalysts 846 market contain fluorine.1d Fluorine is the most electronegative 2.1.5. One-Pot and Tandem Processes 848 element in the periodic table, and the introduction of one or 2.2. Nucleophilic Fluorination 850 more fluorine atoms into a molecule can result in greatly 3. Catalytic Enantioselective Trifluoromethylation perturbed properties. Fluorine substituents can potentially and Perfluoroalkylation 853 impact a number of variables, such as the acidity or basicity of 3.1. Asymmetric Nucleophilic Trifluoromethyla- neighboring groups, dipole moment, and properties such as tion 853 lipophilicity, metabolic stability, and bioavailability. The 3.1.1. Overview of Nucleophilic Trifluorome- multitude of effects that can arise from the introduction of thylation 853 fluorine in small molecules in the context of medicinal 3.1.2. -

CV-PSB-August 2020
Curriculum Vitae Phil S. Baran Appointment: Scripps Research Professor, Department of Chemistry 10550 North Torrey Pines Road, BCC-436 La Jolla, California 92037 Telephone: (858) 784-7373 Facsimile: (858) 784-7575 Email: [email protected] Website: www.scripps.edu/chem/baran/ January, 2013 Darlene Shiley Professor of Chemistry April, 2009 Member, Skaggs Institute for Chemical Biology June, 2008 Professor of Chemistry July, 2006 Associate Professor of Chemistry (with Tenure) June, 2003 Assistant Professor of Chemistry Date/Place of Birth: 10 Aug 1977 / Denville, NJ, USA Citizenship: United States Education 2001 – 2003 Postdoctoral Associate Advisor: Professor E.J. Corey Harvard University, Cambridge, Massachusetts 1997 – 2001 Ph.D. Graduate Student in Chemistry Advisor: Professor K.C. Nicolaou The Scripps Research Institute, La Jolla, California 1995 – 1997 B.S. with Honors in Chemistry Advisor: Professor D.I. Schuster New York University, New York, New York 1991 – 1995 Simultaneous high school graduation from Mt. Dora High School and A.A. degree with honors, Lake Sumter Community College, Florida Awards • Inhoffen Medal, 2019 • Manchot Research Professorship, 2017 • Member, The National Academy of Sciences, 2017 • Emanuel Merck Lectureship, 2017 • Blavatnik National Laureate in Chemistry, 2016 • ACS Elias J. Corey Award, 2016 • Member, American Academy of Arts and Sciences, 2015 • College of Arts and Science Alumni Distinguished Service Award, New York University, 2015 • Reagent of the Year Award (EROS), 2015 • Mukaiyama Award, 2014 • -

Development of Palladium-Catalyzed Decarboxylative Allylation of Electron-Deficient Sulfones: Method Development and Mechanistic Studies
Development of Palladium-Catalyzed Decarboxylative Allylation of Electron-Deficient Sulfones: Method Development and Mechanistic Studies by Monica Anne Gill A thesis submitted to the Faculty of Graduate and Postdoctoral Affairs in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry Carleton University Ottawa, Ontario © 2015, Monica Anne Gill Abstract Palladium-catalyzed decarboxylative allylation is a powerful method of carbon-carbon bond construction. This methodology relies on an electron- withdrawing group to promote the reaction. The use of sulfones in decarboxylative allylation has been explored using the trifluoromethylsulfonyl (triflyl) group, as well as the bis(3,5-trifluoromethyl)phenylsulfonyl (BTMP) group. These substrates are highly reactive at room temperature (triflyl) and 50 oC (BTMP sulfones). A detailed mechanistic study using deuterium-labelled substrates was performed to understand the origin of the protonation side-product. It was proposed that a β- hydride elimination from the η1 allyl on palladium could generate a palladium hydride intermediate along with an allene. Although small amounts of deuterium incorporation were observed in the protonated products, the proposed mechanism could not be the major pathway. Using isotopically labelled ligand, however, all protonation was supressed. This suggests that the origin of the proton is actually from the ligand and that kinetic isotope effects may be responsible for inhibiting the protonation pathway with labelled ligand. ii Acknowledgments I would like to thank Dr. Jeff Manthorpe for giving me the chance to pursue my PhD in his lab. I always appreciated Jeff’s enthusiasm for organic chemistry as well as his vast knowledge of the subject. -
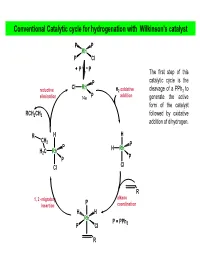
Conventional Catalytic Cycle for Hydrogenation with Wilkinson's Catalyst
Conventional Catalytic cycle for hydrogenation with Wilkinson’s catalyst P P Rh P Cl PP The first step of this P catalytic cycle is the Cl Rh reductive H2 oxidative cleavage of a PPh3 to elimination 14e P addition generate the active form of the catalyst RCH2CH3 followed by oxidative addition of dihydrogen. R H H CH 2 P P H Rh H2C Rh P P Cl Cl R alkene 1, 2 -migratory P insertion coordination H H Rh P = PPh P Cl 3 R AJELIAS L7-S18 catalytic cycle for hydrogenation H H2 oxidative P P addition H P Kinetic studies have Rh Rh shown that the P Cl P Cl dissociation of PPh3 P from the distorted P square planar complex PP (due to trans RhCl(PPh3)3 in effect of H ) H benzene occurs only to a very small extent H2 oxidative P (k = 2.3 × 10–7 M at P addition HRh Cl Rh 25°C), and P P under an atmosphere Cl of H2, a solution of RCH2CH3 RhCl(PPh3)3 becomes reductive alkene yellow as a result of elimination the oxidative addition of H2 to give cis- R H P CH2 H2RhCl(PPh3)3. P H H H2CRh Rh P 1, 2 -migratory P Cl insertion Cl R The trans effect is the labilization (making unstable) of ligands that are trans to certain other ligands, which can thus be regarded as trans-directing ligands. The intensity of the trans effect (as measured by the increase in rate of substitution of the trans ligand) follows this sequence: H2O, OH− < NH3 < py < Cl− < Br− < I−, < PR3, CH3− < H−, NO, CO AJELIAS L7-S19 Relative reactivity of alkenes for homogenous catalytic hydrogenation • Cis alkenes undergo hydrogenation more readily than trans alkenes •Internal and branched alkenes -

Basics of Catalysis and Kinetics
Basics of Catalysis and Kinetics Nobel laureates in catalysis: Haber (1918) Ziegler and Natta (1963) Wilkinson, Fischer (1973) Knowles, Noyori, Sharpless (2001) Grubbs, Schrock, Chauvin (2006) Ertl (2007) Heck, Negishi, Suzuki (2010) G. Poli Definition The concept of catalyst was first introduced by Berzelius in the 19th century. Subsequently, Ostwald came up with the definition that we still use today: “A catalyst is a substance that increases the rate of a chemical reaction, without being consumed or produced”… Berzelius, J. J. Ann. Chim. Phys. 1836, 61, 146-151 Ostwald, W. Z. Phys. Chem. 1894, 15, 705-706 G. Poli The Catalyst and the Rate of a Reaction ΔG non catalyzed catalyzed A B Reaction coordinate The catalyst changes only the rate of the reaction, and does not change the thermodynamics. However, the temperature can affect equilibrium concentrations: ΔG = ΔH - T ΔS G. Poli Historical basis for the Analysis of Catalytic Kinetics The Michaelis-Menten Kinetics G. Poli Thermal, Catalyzed, Inhibited, and Promoted Reactions I II III IV uncatalyzed catalysis inhibition stoichimetric process (no catalysis) promotion (no catalysis) A A A A Acat Bcat Ainhib Binhib Aprom B B (+ cat) B Bprom SM is stabilized SM is stabilized more than TS --> a further reaction is less than TS the uncatalyzed path needed to liberate B is preferred G. Poli Thermal, Catalyzed, Inhibited, and Promoted Reactions O NH HN CCl3 Ar O CCl3 xylenes 140°C type I Ar CCl3 CCl3 O NH PdCl2(PhCN)2 (5 mol%) O NH type II THF, rt N H H N B N N N N B Pt N N 1. -

Decomposition of Ruthenium Olefin Metathesis Catalyst
catalysts Review Decomposition of Ruthenium OlefinOlefin Metathesis CatalystMetathesis Catalyst Magdalena Jawiczuk 1,, Anna Anna Marczyk Marczyk 1,21,2 andand Bartosz Bartosz Trzaskowski Trzaskowski 1,* 1,* 1 1 CentreCentre of of New New Technologies, Technologies, University University of of Warsaw, Warsaw, Banacha Banacha 2c, 2c, 02-097 02-097 Warsaw, Warsaw, Poland; [email protected]@cent.uw.edu.pl (M.J.); (M.J.); [email protected] [email protected] (A.M.) (A.M.) 2 Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland 2 Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland * Correspondence: [email protected] * Correspondence: [email protected] Received: 28 28 June 2020; Accepted: 02 2 AugustAugust 2020;2020; Published:Published: 5date August 2020 Abstract: RutheniumRuthenium olefin olefin metathesis metathesis catalysts catalysts are are one one of of the most commonly used class of catalysts. There There are are multiple multiple reviews reviews on on their their us useses in in various branches of chemistry and other sciences but a detailed review of their decomposition is missing, despite a large number of recent and important advances advances in in this this field. field. In In particular, particular, in in the the last last five five years years several several new new mechanism mechanism of decomposition,of decomposition, both both olefin-driven olefin-driven as well as well as induc as induceded by external by external agents, agents, have have been been suggested suggested and usedand usedto explain to explain differences differences in the decomposition in the decomposition rates and rates the metathesis and the metathesis activities activitiesof both standard, of both N-heterocyclicstandard, N-heterocyclic carbene-based carbene-based systems and systems the recently and the developed recently developed cyclic alkyl cyclic amino alkyl carbene- amino containingcarbene-containing complexes.