Mechanistic Aspects of the Kharasch Addition Reaction Catalyzed by Organonickel(II)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry
REVIEWS Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry Eiichi Nakamura* and Seiji Mori Organocopper reagents provide the principles. This review will summarize example of molecular recognition and most general synthetic tools in organic first the general structural features of supramolecular chemistry, which chemistry for nucleophilic delivery of organocopper compounds and the pre- chemists have long exploited without hard carbanions to electrophilic car- vious mechanistic arguments, and then knowing it. Reasoning about the bon centers. A number of structural describe the most recent mechanistic uniqueness of the copper atom among and mechanistic studies have been pictures obtained through high-level neighboring metal elements in the reported and have led to a wide variety quantum mechanical calculations for periodic table will be presented. of mechanistic proposals, some of three typical organocuprate reactions, which might even be contradictory to carbocupration, conjugate addition, Keywords: catalysis ´ conjugate addi- others. With the recent advent of and SN2 alkylation. The unified view tions ´ copper ´ density functional physical and theoretical methodolo- on the nucleophilic reactivities of met- calculations ´ supramolecular chemis- gies, the accumulated knowledge on al organocuprate clusters thus ob- try organocopper chemistry is being put tained has indicated that organocup- together into a few major mechanistic rate chemistry represents an intricate 1. Introduction 1 R Cu X The desire to learn about the nature of elements has been R or R1 R and will remain a main concern of chemists. In this review, we R Cu will consider what properties of copper make organocopper R1 chemistry so useful in organic chemistry. -

Mechanistic Considerations on the Hydrodechlorination Process of Polychloroarenes
Chapter 3 Mechanistic Considerations on the Hydrodechlorination Process of Polychloroarenes YoshiharuYoshiharu Mitoma, Mitoma, Yumi KatayamaYumi Katayama andand CristianCristian Simion Simion Additional information is available at the end of the chapter http://dx.doi.org/10.5772/intechopen.79083 Abstract Defunctionalization of organochlorines through reductive dechlorination (also known as hydrodechlorination—replacement of chlorine atoms by hydrogen—is one of the main methodologies used in the detoxification of these harmful compounds. Most of the pub- lished papers on this particular matter focused on specific reagents, reaction conditions, and mainly result efficiency. Some of the authors were also concerned with reaction -path ways (e.g., the order in which chlorine atoms were removed from a polychlorinated aro- matic substrate—polychlorinated biphenyls, PCBs; polychlorinated dibenzo-p-dioxins, PCDDs; or polychlorinated dibenzofurans, PCDFs). However, the papers that dealt with the investigation of reaction mechanism were rather scarce. This chapter presents the advances made by researchers in understanding, from a mechanistic point of view, the hydrodechlorination process, along with our own assumptions. In doing so, it would be easier to predict the behavior of such compounds in a specific environment, showing more clearly the scope and limitations of each process, depending on the reaction condi- tions and reagents. Keywords: hydrodechlorination, reaction mechanism, metal/hydrogen donor 1. Introduction Most chlorinated and especially polychlorinated arenes (such as polychloro-dibenzo-p-diox- ins 1, polychloro-dibenzofurans 2, or polychlorobiphenyls 3) are persistent organic pollutants (POPs) that are harmful to both man and the environment [1–5]. © 2016 The Author(s). Licensee InTech. This chapter is distributed under the terms of the Creative Commons © 2018 The Author(s). -

Recent Advances on Mechanistic Studies on C–H Activation
Open Chem., 2018; 16: 1001–1058 Review Article Open Access Daniel Gallego*, Edwin A. Baquero Recent Advances on Mechanistic Studies on C–H Activation Catalyzed by Base Metals https:// doi.org/10.1515/chem-2018-0102 received March 26, 2018; accepted June 3, 2018. 1Introduction Abstract: During the last ten years, base metals have Application in organic synthesis of transition metal- become very attractive to the organometallic and catalytic catalyzed cross coupling reactions has been positioned community on activation of C-H bonds for their catalytic as one of the most important breakthroughs during the functionalization. In contrast to the statement that new millennia. The seminal works based on Pd–catalysts base metals differ on their mode of action most of the in the 70’s by Heck, Noyori and Suzuki set a new frontier manuscripts mistakenly rely on well-studied mechanisms between homogeneous catalysis and synthetic organic for precious metals while proposing plausible chemistry [1-5]. Late transition metals, mostly the precious mechanisms. Consequently, few literature examples metals, stand as the most versatile catalytic systems for a are found where a thorough mechanistic investigation variety of functionalization reactions demonstrating their have been conducted with strong support either by robustness in several applications in organic synthesis [6- theoretical calculations or experimentation. Therefore, 12]. Owing to the common interest in the catalysts mode we consider of highly scientific interest reviewing the of action by many research groups, nowadays we have a last advances on mechanistic studies on Fe, Co and Mn wide understanding of the mechanistic aspects of precious on C-H functionalization in order to get a deep insight on metal-catalyzed reactions. -

2.1 the 18-ELECTRON RULE the 18E Rule1 Is a Way to Help Us Decide
30 GENERAL PROPERTIES OF ORGANOMETALLIC COMPLEXES 2.1 THE 18-ELECTRON RULE The 18e rule1 is a way to help us decide whether a given d-block transition metal organometallic complex is likely to be stable. Not all the organic formulas we can write down correspond to stable species. For example, CH5 requires a 5-valent carbon and is therefore not stable. Stable compounds, such as CH4,have the noble gas octet, and so carbon can be thought of as following an 8e rule. This corresponds to carbon using its s and three p orbitals to form four filled bonding orbitals and four unfilled antibonding orbitals. On the covalent model, we can consider that of the eight electrons required to fill the bonding orbitals, four come from carbon and one each comes from the four H substituents. We can therefore think of each H atom as being a 1e ligand to carbon. To assign a formal oxidation state to carbon in an organic molecule, we impose an ionic model by artificially dissecting it into ions. Each electron pair in any bond is assigned to the most electronegative of the two atoms or groups that constitute the bond. For methane, this dissection gives C4− + 4H+, with carbon as the more electronegative element. This makes methane an 8e compound with an oxidation state of −4, usually written C(-IV). Note that the net electron count always remains the same, whether we adopt the covalent (4e {Catom}+4 × 1e {4H atoms}=8e) or ionic (8e{C4−ion}+4 × 0e{4H+ions}=8e) model. -

Assessing the Feasibility of a One-Pot, Tandem Olefin Metathesis and Isomerization Sequence to Synthesize Conjugated Aromatic Olefins
University of Tennessee at Chattanooga UTC Scholar Student Research, Creative Works, and Honors Theses Publications 8-2016 Assessing the feasibility of a one-pot, tandem olefin metathesis and isomerization sequence to synthesize conjugated aromatic olefins Ajay D. Makwana University of Tennessee at Chattanooga, [email protected] Follow this and additional works at: https://scholar.utc.edu/honors-theses Part of the Chemistry Commons Recommended Citation Makwana, Ajay D., "Assessing the feasibility of a one-pot, tandem olefin metathesis and isomerization sequence to synthesize conjugated aromatic olefins" (2016). Honors Theses. This Theses is brought to you for free and open access by the Student Research, Creative Works, and Publications at UTC Scholar. It has been accepted for inclusion in Honors Theses by an authorized administrator of UTC Scholar. For more information, please contact [email protected]. Assessing the Feasibility of a One-Pot, Tandem Olefin Metathesis and Isomerization Sequence to Synthesize Conjugated Aromatic Olefins Ajay D. Makwana Departmental Honors Thesis The University of Tennessee at Chattanooga Department of Chemistry Project Director: Dr. Kyle S. Knight Examination Date: April 4, 2016 Members of Examination Committee: Dr. John P. Lee Dr. Han J. Park Dr. David Giles 1 Abstract The synthesis of substituted phenylpropene dimers using a one-pot, tandem olefin metathesis and isomerization sequence has been studied. This sequence relies on the facilitated, in-situ conversion of a ruthenium carbene species (Ru=C) to a ruthenium hydride species (Ru-H) upon addition of an inorganic hydride source. Three separate reactions occur within one reaction flask: 1) olefin metathesis of the starting phenylpropene to yield phenylpropene dimer via Ru=C catalyst, 2) conversion of Ru=C to Ru-H via addition of an inorganic hydride source, 3) isomerization of phenylpropene dimer via insertion and β-hydride elimination to yield conjugated product. -

Wilkinson's Catalyst
Homogeneous Catalytic Processes Hydroformylation RCH2CH2 O RCH CH2 + CO + H2 Co(I), Rh(I) or Pt(II) H Oxidation H3C O H2CCH2 + O2 Pd(II) or Cu(II) H Carbonylation H3C O - CH3OH + CO [RhI2(CO)2] OH Hydrocyanation [Ni{P(OR) } ] H CCC CH + 2HCN 3 4 NCCH CH CH CH CN 2 H H 2 2 2 2 2 Cyclotrimerization Ni(acac) 3 HC CH 2 Hydrogenation of Alkenes The most commonly used catalyst is the Wilkinson’s Catalyst Many alkenes are hydrogenated with hydrogen at 1atm pressure or less . Wilkinson’s catalyst is highly sensitive to the nature of the phosphine ligand and the alkene substrate. Analogous catalysts with alkyl phosphine ligands are inactive Highly hindered alkenes and ethylene are not hydrogenated by the catalyst Wilkinson’s Catalyst Ph3P PPh3 R CH3 Rh H2 Ph3P Cl Reductive Elimination Oxidative Addition R H H PPh3 H PPh3 Rh Rh Ph3P Cl Ph3P Cl PPh3 PPh3 Ligand Dissociation Ligand Association PPh3 PPh3 H R H PPh3 Rh H PPh3 Cl Rh Ph3P Cl PPh3 H Migratory Insertion H PPh3 Rh R Ph3P Cl Alkene Coordination R Hydroformylation Catalyst + CO + H2 RCH2CH2CHO R A less common,,pppyy but more appropriate name is hydrocarbonylation Both cobalt and rhodium complexes are used as catalysts. Alkene iitiisomerization, alkene hdhydrogena tion and ftiformation of bhdbranched alde hy des are the possible side reactions. Cobalt catalysts operate at 150 ºC and 250 atm, whereas Rhodium catalysts operate at moderate temperatures and 1 atm. Rhodium catalysts promotes the formation of linear aldehydes. Cobalt catalysts do so if modified with alkylphosphine ligands. -

Neutral, Cationic and Anionic Organonickel and -Palladium Complexes Supported by Cite This: Dalton Trans., 2020, 49, 322 Iminophosphine/Phosphinoenaminato Ligands†‡
Dalton Transactions View Article Online PAPER View Journal | View Issue Neutral, cationic and anionic organonickel and -palladium complexes supported by Cite this: Dalton Trans., 2020, 49, 322 iminophosphine/phosphinoenaminato ligands†‡ Tomás G. Santiago, Carmen Urbaneja, Eleuterio Álvarez, Elena Ávila, Pilar Palma and Juan Cámpora * We report a series of organometallic nickel and palladium complexes containing iminophosphine ligands R2PCH2C(Ph) = N-Dipp (Dipp = 2,6-diisopropylphenyl; R = iPr, La; R = Ph, Lb; and R = o-C6H4OMe, Lc), synthesized by ligand exchange or oxidative addition reactions, and we investigate the capacity of such ligands to undergo reversible deprotonation to the corresponding phosphinoenaminato species. In the attempted ligand exchange reaction of the nickel bis(trimethylsilyl)methyl precursor [Ni(CH2SiMe3)2Py2] with Lb, the iminophosphine acts as a weak acid rather than a neutral ligand, cleaving one of the Ni–C bonds, to afford the phosphinoenaminato complex [Ni(CH2SiMe3)(L’b)(Py)] (L’b = conjugate base of Lb). Creative Commons Attribution-NonCommercial 3.0 Unported Licence. + We disclose a general method for the syntheses of complexes [Ni(CH2SiMe3)(L)(Py)] (L = La, Lb or Lc), and demonstrate that iminophosphine deprotonation is a general feature and occurs reversibly in the coordination sphere of the metal. By studying proton exchange reactions of the cation [Ni(CH2SiMe3)(Lb) (Py)]+ with bases of different strength we show that the conjugate phosphinoenaminato ligand in Received 17th October 2019, [Ni(CH2SiMe3)(L’b)(Py)] is a base with strength comparable to DBU in THF. The acyl group in the function- Accepted 19th November 2019 alized aryl complex [Ni(p-C6H4COCH3)(Br)(La)] does not interfere in the iminophosphine deprotonation with DOI: 10.1039/c9dt04062e − + NaH. -

Title Investigation of Organoiron Catalysis in Kumada‒Tamao‒Corriu-Type Cross-Coupling Reaction Assisted by Solution-Phase X
Investigation of Organoiron Catalysis in Title Kumada‒Tamao‒Corriu-Type Cross-Coupling Reaction Assisted by Solution-Phase X-ray Absorption Spectroscopy Takaya, Hikaru; Nakajima, Sho; Nakagawa, Naohisa; Isozaki, Katsuhiro; Iwamoto, Takahiro; Imayoshi, Ryuji; Gower, Nicholas J.; Adak, Laksmikanta; Hatakeyama, Takuji; Honma, Author(s) Tetsuo; Takagaki, Masafumi; Sunada, Yusuke; Nagashima, Hideo; Hashizume, Daisuke; Takahashi, Osamu; Nakamura, Masaharu Bulletin of the Chemical Society of Japan (2015), 88(3): 410- Citation 418 Issue Date 2015 URL http://hdl.handle.net/2433/196165 © 2015 The Chemical Society of Japan.; 本文ファイルは出版 社の許可を得て登録しています.; This is not the published Right version. Please cite only the published version.; この論文は出 版社版でありません。引用の際には出版社版をご確認ご 利用ください。 Type Journal Article Textversion author Kyoto University Bull. Chem. Soc. Jpn. in press (doi:10.1246/bcsj.20140376) Graphical Abstract Investigation of Organoiron Catalysis in Kumada–Tamao–Corriu-Type Cross-Coupling Reaction Assisted by Solution-Phase X-ray Absorption Spectroscopy H. Takaya, S. Nakajima, N. Nakagawa, K. Isozaki, T. Iwamoto, R. Imayoshi, N. J. Gower, L. Adak, T. Hatakeyama, T. Honma, M. Takagaki, Y. Sunada, H. Nagashima, D. Hashizume, O. Takahashi, and M. Nakamura Solution-phase molecular structures of organoiron intermediates of Kumada–Tamao–Corriu-type cross-coupling were illuminated by X-ray absorption spectroscopy. The intermediacy of halomesityl iron complex of II II Fe BrMes(SciOPP) and dimesityl iron complex of Fe Mes2(SciOPP) was adequately elucidated with formal non- redox FeII/FeII catalytic cycle. 1 Investigation of Organoiron Catalysis in Kumada–Tamao–Corriu-Type Cross-Coupling Reaction Assisted by Solution-Phase X-ray Absorption Spectroscopy Hikaru Takaya,*a,b Sho Nakajima,a,b Naohisa Nakagawa,a Katsuhiro Isozaki,a,b,g Takahiro Iwamoto,a,b,g Ryuji Imayoshi,a,b Nicholas J. -
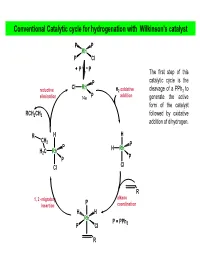
Conventional Catalytic Cycle for Hydrogenation with Wilkinson's Catalyst
Conventional Catalytic cycle for hydrogenation with Wilkinson’s catalyst P P Rh P Cl PP The first step of this P catalytic cycle is the Cl Rh reductive H2 oxidative cleavage of a PPh3 to elimination 14e P addition generate the active form of the catalyst RCH2CH3 followed by oxidative addition of dihydrogen. R H H CH 2 P P H Rh H2C Rh P P Cl Cl R alkene 1, 2 -migratory P insertion coordination H H Rh P = PPh P Cl 3 R AJELIAS L7-S18 catalytic cycle for hydrogenation H H2 oxidative P P addition H P Kinetic studies have Rh Rh shown that the P Cl P Cl dissociation of PPh3 P from the distorted P square planar complex PP (due to trans RhCl(PPh3)3 in effect of H ) H benzene occurs only to a very small extent H2 oxidative P (k = 2.3 × 10–7 M at P addition HRh Cl Rh 25°C), and P P under an atmosphere Cl of H2, a solution of RCH2CH3 RhCl(PPh3)3 becomes reductive alkene yellow as a result of elimination the oxidative addition of H2 to give cis- R H P CH2 H2RhCl(PPh3)3. P H H H2CRh Rh P 1, 2 -migratory P Cl insertion Cl R The trans effect is the labilization (making unstable) of ligands that are trans to certain other ligands, which can thus be regarded as trans-directing ligands. The intensity of the trans effect (as measured by the increase in rate of substitution of the trans ligand) follows this sequence: H2O, OH− < NH3 < py < Cl− < Br− < I−, < PR3, CH3− < H−, NO, CO AJELIAS L7-S19 Relative reactivity of alkenes for homogenous catalytic hydrogenation • Cis alkenes undergo hydrogenation more readily than trans alkenes •Internal and branched alkenes -

Basics of Catalysis and Kinetics
Basics of Catalysis and Kinetics Nobel laureates in catalysis: Haber (1918) Ziegler and Natta (1963) Wilkinson, Fischer (1973) Knowles, Noyori, Sharpless (2001) Grubbs, Schrock, Chauvin (2006) Ertl (2007) Heck, Negishi, Suzuki (2010) G. Poli Definition The concept of catalyst was first introduced by Berzelius in the 19th century. Subsequently, Ostwald came up with the definition that we still use today: “A catalyst is a substance that increases the rate of a chemical reaction, without being consumed or produced”… Berzelius, J. J. Ann. Chim. Phys. 1836, 61, 146-151 Ostwald, W. Z. Phys. Chem. 1894, 15, 705-706 G. Poli The Catalyst and the Rate of a Reaction ΔG non catalyzed catalyzed A B Reaction coordinate The catalyst changes only the rate of the reaction, and does not change the thermodynamics. However, the temperature can affect equilibrium concentrations: ΔG = ΔH - T ΔS G. Poli Historical basis for the Analysis of Catalytic Kinetics The Michaelis-Menten Kinetics G. Poli Thermal, Catalyzed, Inhibited, and Promoted Reactions I II III IV uncatalyzed catalysis inhibition stoichimetric process (no catalysis) promotion (no catalysis) A A A A Acat Bcat Ainhib Binhib Aprom B B (+ cat) B Bprom SM is stabilized SM is stabilized more than TS --> a further reaction is less than TS the uncatalyzed path needed to liberate B is preferred G. Poli Thermal, Catalyzed, Inhibited, and Promoted Reactions O NH HN CCl3 Ar O CCl3 xylenes 140°C type I Ar CCl3 CCl3 O NH PdCl2(PhCN)2 (5 mol%) O NH type II THF, rt N H H N B N N N N B Pt N N 1. -

Decomposition of Ruthenium Olefin Metathesis Catalyst
catalysts Review Decomposition of Ruthenium OlefinOlefin Metathesis CatalystMetathesis Catalyst Magdalena Jawiczuk 1,, Anna Anna Marczyk Marczyk 1,21,2 andand Bartosz Bartosz Trzaskowski Trzaskowski 1,* 1,* 1 1 CentreCentre of of New New Technologies, Technologies, University University of of Warsaw, Warsaw, Banacha Banacha 2c, 2c, 02-097 02-097 Warsaw, Warsaw, Poland; [email protected]@cent.uw.edu.pl (M.J.); (M.J.); [email protected] [email protected] (A.M.) (A.M.) 2 Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland 2 Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland * Correspondence: [email protected] * Correspondence: [email protected] Received: 28 28 June 2020; Accepted: 02 2 AugustAugust 2020;2020; Published:Published: 5date August 2020 Abstract: RutheniumRuthenium olefin olefin metathesis metathesis catalysts catalysts are are one one of of the most commonly used class of catalysts. There There are are multiple multiple reviews reviews on on their their us useses in in various branches of chemistry and other sciences but a detailed review of their decomposition is missing, despite a large number of recent and important advances advances in in this this field. field. In In particular, particular, in in the the last last five five years years several several new new mechanism mechanism of decomposition,of decomposition, both both olefin-driven olefin-driven as well as well as induc as induceded by external by external agents, agents, have have been been suggested suggested and usedand usedto explain to explain differences differences in the decomposition in the decomposition rates and rates the metathesis and the metathesis activities activitiesof both standard, of both N-heterocyclicstandard, N-heterocyclic carbene-based carbene-based systems and systems the recently and the developed recently developed cyclic alkyl cyclic amino alkyl carbene- amino containingcarbene-containing complexes. -

X International Conference “Mechanisms of Catalytic Reactions”
Boreskov Institute of Catalysis SB RAS, Novosibirsk, Russia Zelinsky Institute of Organic Chemistry RAS, Moscow, Russia Lomonosov Moscow State University, Moscow, Russia 2016 X International Conference “Mechanisms of Catalytic Reactions” Svetlogorsk, Kaliningrad Region, Russia October 2 - 6, 2016 ABSTRACTS Novosibirsk-2016 Boreskov Institute of Catalysis SB RAS, Novosibirsk, Russia Zelinsky Institute of Organic Chemistry RAS, Moscow, Russia Lomonosov Moscow State University, Moscow, Russia X International Conference “Mechanisms of Catalytic Reactions” Svetlogorsk, Kaliningrad Region, Russia October 2 - 6, 2016 ABSTRACTS Novosibirsk-2016 УДК 544.47+66.09 ББК Г544 M45 Mechanisms of Catalytic Reactions. X International Conference (MCR-X). (October 2 - 6, 2016, Svetlogorsk, Kaliningrad Region, Russia) [Electronic resourse]: Book of abstracts / Boreskov Institute of Catalysis SB RAS ed.: prof. V.I. Bukhtiyarov, - Novosibirsk: BIC, 2016. p.328, – 1 electronic optical disc (CD-R). ISBN 978-5-906376-15-2 В надзаг.: Boreskov Institute of Catalysis SB RAS, Novosibirsk, Russia Zelinsky Institute of Organic Chemistry RAS, Moscow, Russia Lomonosov Moscow State University, Moscow, Russia Topics of book: – First-principles approach, theory and simulation in catalysis; – Advanced methods for studies of mechanisms of catalyzed reactions; – In-situ and operando studies of model and real catalysts; – Kinetics and reaction intermediates of catalyzed processes; – From mechanistic studies to design of advanced catalyst systems. The Conference is accompanied