2.1 the 18-ELECTRON RULE the 18E Rule1 Is a Way to Help Us Decide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recent Advances on Mechanistic Studies on C–H Activation
Open Chem., 2018; 16: 1001–1058 Review Article Open Access Daniel Gallego*, Edwin A. Baquero Recent Advances on Mechanistic Studies on C–H Activation Catalyzed by Base Metals https:// doi.org/10.1515/chem-2018-0102 received March 26, 2018; accepted June 3, 2018. 1Introduction Abstract: During the last ten years, base metals have Application in organic synthesis of transition metal- become very attractive to the organometallic and catalytic catalyzed cross coupling reactions has been positioned community on activation of C-H bonds for their catalytic as one of the most important breakthroughs during the functionalization. In contrast to the statement that new millennia. The seminal works based on Pd–catalysts base metals differ on their mode of action most of the in the 70’s by Heck, Noyori and Suzuki set a new frontier manuscripts mistakenly rely on well-studied mechanisms between homogeneous catalysis and synthetic organic for precious metals while proposing plausible chemistry [1-5]. Late transition metals, mostly the precious mechanisms. Consequently, few literature examples metals, stand as the most versatile catalytic systems for a are found where a thorough mechanistic investigation variety of functionalization reactions demonstrating their have been conducted with strong support either by robustness in several applications in organic synthesis [6- theoretical calculations or experimentation. Therefore, 12]. Owing to the common interest in the catalysts mode we consider of highly scientific interest reviewing the of action by many research groups, nowadays we have a last advances on mechanistic studies on Fe, Co and Mn wide understanding of the mechanistic aspects of precious on C-H functionalization in order to get a deep insight on metal-catalyzed reactions. -

Assessing the Feasibility of a One-Pot, Tandem Olefin Metathesis and Isomerization Sequence to Synthesize Conjugated Aromatic Olefins
University of Tennessee at Chattanooga UTC Scholar Student Research, Creative Works, and Honors Theses Publications 8-2016 Assessing the feasibility of a one-pot, tandem olefin metathesis and isomerization sequence to synthesize conjugated aromatic olefins Ajay D. Makwana University of Tennessee at Chattanooga, [email protected] Follow this and additional works at: https://scholar.utc.edu/honors-theses Part of the Chemistry Commons Recommended Citation Makwana, Ajay D., "Assessing the feasibility of a one-pot, tandem olefin metathesis and isomerization sequence to synthesize conjugated aromatic olefins" (2016). Honors Theses. This Theses is brought to you for free and open access by the Student Research, Creative Works, and Publications at UTC Scholar. It has been accepted for inclusion in Honors Theses by an authorized administrator of UTC Scholar. For more information, please contact [email protected]. Assessing the Feasibility of a One-Pot, Tandem Olefin Metathesis and Isomerization Sequence to Synthesize Conjugated Aromatic Olefins Ajay D. Makwana Departmental Honors Thesis The University of Tennessee at Chattanooga Department of Chemistry Project Director: Dr. Kyle S. Knight Examination Date: April 4, 2016 Members of Examination Committee: Dr. John P. Lee Dr. Han J. Park Dr. David Giles 1 Abstract The synthesis of substituted phenylpropene dimers using a one-pot, tandem olefin metathesis and isomerization sequence has been studied. This sequence relies on the facilitated, in-situ conversion of a ruthenium carbene species (Ru=C) to a ruthenium hydride species (Ru-H) upon addition of an inorganic hydride source. Three separate reactions occur within one reaction flask: 1) olefin metathesis of the starting phenylpropene to yield phenylpropene dimer via Ru=C catalyst, 2) conversion of Ru=C to Ru-H via addition of an inorganic hydride source, 3) isomerization of phenylpropene dimer via insertion and β-hydride elimination to yield conjugated product. -

Wilkinson's Catalyst
Homogeneous Catalytic Processes Hydroformylation RCH2CH2 O RCH CH2 + CO + H2 Co(I), Rh(I) or Pt(II) H Oxidation H3C O H2CCH2 + O2 Pd(II) or Cu(II) H Carbonylation H3C O - CH3OH + CO [RhI2(CO)2] OH Hydrocyanation [Ni{P(OR) } ] H CCC CH + 2HCN 3 4 NCCH CH CH CH CN 2 H H 2 2 2 2 2 Cyclotrimerization Ni(acac) 3 HC CH 2 Hydrogenation of Alkenes The most commonly used catalyst is the Wilkinson’s Catalyst Many alkenes are hydrogenated with hydrogen at 1atm pressure or less . Wilkinson’s catalyst is highly sensitive to the nature of the phosphine ligand and the alkene substrate. Analogous catalysts with alkyl phosphine ligands are inactive Highly hindered alkenes and ethylene are not hydrogenated by the catalyst Wilkinson’s Catalyst Ph3P PPh3 R CH3 Rh H2 Ph3P Cl Reductive Elimination Oxidative Addition R H H PPh3 H PPh3 Rh Rh Ph3P Cl Ph3P Cl PPh3 PPh3 Ligand Dissociation Ligand Association PPh3 PPh3 H R H PPh3 Rh H PPh3 Cl Rh Ph3P Cl PPh3 H Migratory Insertion H PPh3 Rh R Ph3P Cl Alkene Coordination R Hydroformylation Catalyst + CO + H2 RCH2CH2CHO R A less common,,pppyy but more appropriate name is hydrocarbonylation Both cobalt and rhodium complexes are used as catalysts. Alkene iitiisomerization, alkene hdhydrogena tion and ftiformation of bhdbranched alde hy des are the possible side reactions. Cobalt catalysts operate at 150 ºC and 250 atm, whereas Rhodium catalysts operate at moderate temperatures and 1 atm. Rhodium catalysts promotes the formation of linear aldehydes. Cobalt catalysts do so if modified with alkylphosphine ligands. -
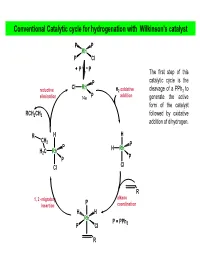
Conventional Catalytic Cycle for Hydrogenation with Wilkinson's Catalyst
Conventional Catalytic cycle for hydrogenation with Wilkinson’s catalyst P P Rh P Cl PP The first step of this P catalytic cycle is the Cl Rh reductive H2 oxidative cleavage of a PPh3 to elimination 14e P addition generate the active form of the catalyst RCH2CH3 followed by oxidative addition of dihydrogen. R H H CH 2 P P H Rh H2C Rh P P Cl Cl R alkene 1, 2 -migratory P insertion coordination H H Rh P = PPh P Cl 3 R AJELIAS L7-S18 catalytic cycle for hydrogenation H H2 oxidative P P addition H P Kinetic studies have Rh Rh shown that the P Cl P Cl dissociation of PPh3 P from the distorted P square planar complex PP (due to trans RhCl(PPh3)3 in effect of H ) H benzene occurs only to a very small extent H2 oxidative P (k = 2.3 × 10–7 M at P addition HRh Cl Rh 25°C), and P P under an atmosphere Cl of H2, a solution of RCH2CH3 RhCl(PPh3)3 becomes reductive alkene yellow as a result of elimination the oxidative addition of H2 to give cis- R H P CH2 H2RhCl(PPh3)3. P H H H2CRh Rh P 1, 2 -migratory P Cl insertion Cl R The trans effect is the labilization (making unstable) of ligands that are trans to certain other ligands, which can thus be regarded as trans-directing ligands. The intensity of the trans effect (as measured by the increase in rate of substitution of the trans ligand) follows this sequence: H2O, OH− < NH3 < py < Cl− < Br− < I−, < PR3, CH3− < H−, NO, CO AJELIAS L7-S19 Relative reactivity of alkenes for homogenous catalytic hydrogenation • Cis alkenes undergo hydrogenation more readily than trans alkenes •Internal and branched alkenes -

Basics of Catalysis and Kinetics
Basics of Catalysis and Kinetics Nobel laureates in catalysis: Haber (1918) Ziegler and Natta (1963) Wilkinson, Fischer (1973) Knowles, Noyori, Sharpless (2001) Grubbs, Schrock, Chauvin (2006) Ertl (2007) Heck, Negishi, Suzuki (2010) G. Poli Definition The concept of catalyst was first introduced by Berzelius in the 19th century. Subsequently, Ostwald came up with the definition that we still use today: “A catalyst is a substance that increases the rate of a chemical reaction, without being consumed or produced”… Berzelius, J. J. Ann. Chim. Phys. 1836, 61, 146-151 Ostwald, W. Z. Phys. Chem. 1894, 15, 705-706 G. Poli The Catalyst and the Rate of a Reaction ΔG non catalyzed catalyzed A B Reaction coordinate The catalyst changes only the rate of the reaction, and does not change the thermodynamics. However, the temperature can affect equilibrium concentrations: ΔG = ΔH - T ΔS G. Poli Historical basis for the Analysis of Catalytic Kinetics The Michaelis-Menten Kinetics G. Poli Thermal, Catalyzed, Inhibited, and Promoted Reactions I II III IV uncatalyzed catalysis inhibition stoichimetric process (no catalysis) promotion (no catalysis) A A A A Acat Bcat Ainhib Binhib Aprom B B (+ cat) B Bprom SM is stabilized SM is stabilized more than TS --> a further reaction is less than TS the uncatalyzed path needed to liberate B is preferred G. Poli Thermal, Catalyzed, Inhibited, and Promoted Reactions O NH HN CCl3 Ar O CCl3 xylenes 140°C type I Ar CCl3 CCl3 O NH PdCl2(PhCN)2 (5 mol%) O NH type II THF, rt N H H N B N N N N B Pt N N 1. -

Decomposition of Ruthenium Olefin Metathesis Catalyst
catalysts Review Decomposition of Ruthenium OlefinOlefin Metathesis CatalystMetathesis Catalyst Magdalena Jawiczuk 1,, Anna Anna Marczyk Marczyk 1,21,2 andand Bartosz Bartosz Trzaskowski Trzaskowski 1,* 1,* 1 1 CentreCentre of of New New Technologies, Technologies, University University of of Warsaw, Warsaw, Banacha Banacha 2c, 2c, 02-097 02-097 Warsaw, Warsaw, Poland; [email protected]@cent.uw.edu.pl (M.J.); (M.J.); [email protected] [email protected] (A.M.) (A.M.) 2 Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland 2 Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland * Correspondence: [email protected] * Correspondence: [email protected] Received: 28 28 June 2020; Accepted: 02 2 AugustAugust 2020;2020; Published:Published: 5date August 2020 Abstract: RutheniumRuthenium olefin olefin metathesis metathesis catalysts catalysts are are one one of of the most commonly used class of catalysts. There There are are multiple multiple reviews reviews on on their their us useses in in various branches of chemistry and other sciences but a detailed review of their decomposition is missing, despite a large number of recent and important advances advances in in this this field. field. In In particular, particular, in in the the last last five five years years several several new new mechanism mechanism of decomposition,of decomposition, both both olefin-driven olefin-driven as well as well as induc as induceded by external by external agents, agents, have have been been suggested suggested and usedand usedto explain to explain differences differences in the decomposition in the decomposition rates and rates the metathesis and the metathesis activities activitiesof both standard, of both N-heterocyclicstandard, N-heterocyclic carbene-based carbene-based systems and systems the recently and the developed recently developed cyclic alkyl cyclic amino alkyl carbene- amino containingcarbene-containing complexes. -

Chapter 1 the Basic Chemistry of Organopalladium Compounds
Chapter 1 The Basic Chemistry of Organopalladium Compounds 1.1 Characteristic Features of Pd-Promoted or -Catalyzed Reactions There are several features which make reactions involving Pd catalysts and reagents particularly useful and versatile among many transition metals used for organic synthesis. Most importantly, Pd catalysts offer an abundance of possibilities of carbon–carbon bond formation. Importance of the carbon–carbon bond forma- tion in organic synthesis needs no explanation. No other transition metals can offer such versatile methods of the carbon–carbon bond formations as Pd. Tol- erance of Pd catalysts and reagents to many functional groups such as carbonyl and hydroxy groups is the second important feature. Pd-catalyzed reactions can be carried out without protection of these functional groups. Although reactions involving Pd should be carried out carefully, Pd reagents and catalysts are not very sensitive to oxygen and moisture, and even to acid in many reactions catalyzed by Pd–phosphine complexes. It is enough to apply precautions to avoid oxidation of coordinated phosphines, and this can be done easily. On the other hand, the Ni(0) complex is extremely sensitive to oxygen. Of course, Pd is a noble metal and expensive. Its price frequently fluctuates drastically. A few years ago, Pd was more expensive than Pt and Au but cheaper than Rh. As of October 2003, the comparative prices of the noble metals were: Pd (1), Au (1.8), Rh (2.8), Pt (3.3), Ru (0.2). Recently the price of Pd has dropped dramatically, and Pt is currently the most expensive noble metal. Also, the toxicity of Pd has posed no serious problem so far. -

Mechanistic Aspects of the Kharasch Addition Reaction Catalyzed by Organonickel(II)
Organometallics 1997, 16, 4985-4994 4985 Mechanistic Aspects of the Kharasch Addition Reaction Catalyzed by Organonickel(II) Complexes Containing the Monoanionic Terdentate Aryldiamine Ligand System -† [C6H2(CH2NMe2)2-2,6-R-4] Lucia A. van de Kuil,‡,§ David M. Grove,‡ Robert A. Gossage,‡ Jan W. Zwikker,§ Leonardus W. Jenneskens,§ Wiendelt Drenth,§ and Gerard van Koten*,‡ Debye Institute, Departments of Metal-Mediated Synthesis and Physical Organic Chemistry, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands Received January 28, 1997X The addition reaction of polyhalogenated alkanes to alkenes (Kharasch addition reaction) is homogeneously catalyzed in the absence of O2 under mild reaction conditions (25 °C) by II the arylnickel complexes [Ni {C6H2(CH2NMe2)2-2,6-R-4}Br] (R ) H, MeC(O), Cl, MeO, NH2) and shows a high selectivity for the 1:1 adduct. Kinetic data on the catalytic system with [Ni{C6H3(CH2NMe2)2-2,6}Br] (R ) H; abbreviated as [Ni(NCN)Br]), methyl methacrylate, and CCl4 reveal a rate of reaction that is first order in nickel complex and in alkene. In our series of para-substituted arylnickel catalysts, the rate of catalysis increases with the electron donating character of the para substituents on the aryl ligand and this rate correlates directly with the NiII/NiIII redox potential. These data, together with separate spectroscopic studies and results from individual experiments employing other solvents, other polyhalogenated alkanes such as CBr4 and CF3CCl3 and other alkene substrates such as styrene, 1-octene, and cyclohexene, lead to the proposal of a catalytic cycle based on a nonchain mechanism with a mononuclear nickel species. -

Organometallic Catalysis
Organometallic Catalysis • The catalysts we look at are soluble complexes, or homogeneous catalysts, as opposed to catalysts such as palladium on carbon,orheterogeneous catalysts. • These terms are used because the catalyst and substrates for the reaction are in the same phase in the homogeneous, but not in the heterogeneous, type, where catalysis takes place at the surface of a solid catalyst. Catalytic mechanisms are considerably easier to study in homogeneous sy,ystems, where such powerful methods as NMR can be used to both assign structures and follow reaction kinetics. • Homogeneous catalysts have the disadvantage that they can be difficult to separate from the product. • Homogeneous catalysts can also be chemically grafted on to solid supports for greater ease of separation of the catalyst from the reaction products. • Although the catalyst is now technically heterogeneous, it often retains the characteristic reactivity pattern that it showed as a homogeneous catalyst, and its properties are usually distinct from those of any of the classical heterogeneous catalysts—these are sometimes called “heterogenized” homogeneous catalysts. Representative selection of polymer‐bound binary metathesis catalysts. • The mechanistic ideas developed in homogeneous catalysis are also becoming more influential in the field of classical heterogeneous catalysis by suggesting structures for intermediates and mechanisms for reaction steps. • By bringing about a reaction at lower temperature, a catalyst can save energy in commercial applications. It often gives higher selectivity for the desired product, minimizing product separation problems and avoiding the need to discard the undesired product as waste. • Environmental concerns have promoted the idea of atom economy, which values a process most highly when all the atoms in the reagents are used to form the product, minimizing waste. -

Organometallic Compounds
Organometallic Compounds In simpler terms these are compounds containing metal-carbon bonds Examples: CH3-MgBr, Ph-Li, [Ni(CO)4], Ferrocene etc. These compounds can be seen as having covalent bonds between the metal and the carbon atom(s). In general, compounds having a metal-ligand bond of considerable covalent character have similar chemistry and follow the chemical behavior of organometallic compounds Metal-cyano complexes are not considered as organometallic compp,ounds, while metal carbonyl complexes are. 18-electron Rule Having 18 electrons in the outer shell consisting of s, p and d orbitals is considered as an indication of stability as in inert gases. The rule suggests that compounds that can attain 18 electrons within the bonding orbitals of the metal show increased stability Mo contributes 6 electrons Benzene contributes 6 electrons Three CO contribute 6 electrons The compound follows 18-electron rule and is stable Hapticity: The number of atoms in the ligand which are directly coordinated to the metal. Hapticity is denoted as η pentahapto – η5 W contributes 6 electrons One cyclopentadiene contributes 5 electrons The other contributes 3 electrons Two CO contribute 4 electrons trihapto – η3 The compound follows 18-electron rule and is stable 18-electron Rule Metals with odd number of electrons form metal-metal bonds in their carbonyl complexes to satisfy the 18-electron rule CO OC CO CO OC Mn Mn CO OC OC CO OC Carbonyl groups can also bridge between two metals, where they can be seen as contributing one electron each to the two metals Scope of 18-electron rule for d-block organometallic compounds Usually less than Usually 18 16 or 18 18 electrons electrons Sc Ti V Cr Mn Fe Co Ni YZrNb MoTcRu RhPd La Hf Ta W Re Os Ir Pt Oxidation Numbers Oxidation numbers are important in predicting and understanding the reactivity of organometallic compounds. -

LETTER Doi:10.1038/Nature14875
LETTER doi:10.1038/nature14875 Switching on elusive organometallic mechanisms with photoredox catalysis Jack A. Terrett1, James D. Cuthbertson1, Valerie W. Shurtleff1 & David W. C. MacMillan1 Transition-metal-catalysed cross-coupling reactions have become and aryl pseudohalides1,2. Hartwig and colleagues have reported a one of the most used carbon–carbon and carbon–heteroatom Ni(COD)2-catalysed aryl etherification at elevated temperature using bond-forming reactions in chemical synthesis. Recently, nickel three preformed oxides, namely sodium methoxide, ethoxide and catalysis has been shown to participate in a wide variety of C2C tert-butyldimethylsiloxide with electron-deficient aryl bromides26. bond-forming reactions, most notably Negishi, Suzuki–Miyaura, However, a general nickel-catalysed C–O coupling strategy under Stille, Kumada and Hiyama couplings1,2. Despite the tremendous mild conditions has thus far been elusive and no examples with alco- advances in C2C fragment couplings, the ability to forge C2O hols have been documented. We recently questioned whether photo- bonds in a general fashion via nickel catalysis has been largely redox catalysis might be used to modulate the range of nickel alkoxide unsuccessful. The challenge for nickel-mediated alcohol couplings oxidation states that can be accessed during a conventional catalytic has been the mechanistic requirement for the critical C–O bond- cycle. More specifically, we recognized that it should be possible to forming step (formally known as the reductive elimination step) to -

Buchwald-Hartwig Chemistry
Buchwald-Hartwig Chemistry Ian Mangion MacMillan Group Meeting July 30, 2002 ! Historical context ! Development of initial catalytic systems ! Mechanistic studies and rational design ! Reaction Scope Definition of Buchwald-Hartwig Chemistry MLn X + HNR2 NR2 R Base R MLn X + HOR OR R Base R EWG EWG X + MLn R EWG Base R EWG X = I, OTf, Br, Cl M = Pd, Ni, Cu ! Over 70 publications from Buchwald ! Over 50 publications from Hartwig ! Several comprehensive reviews Buchwald, S.; Muci, A. Top. Curr. Chem. 2002; 219 , 133-209 Hartwig, J. Pure Appl. Chem. 1999, 71, 1417 Buchwald, S; Yang, B. J. Orgmet. Chem. 1999, 576, 125 Hartwig, J.; ACIEE. 1998, 37, 2046 Hartwig, J . Acc. Chem. Res . 1998, 31, 852 Buchwald et al. Acc. Chem. Res. 1998, 31, 805 Aryl Aminations Before Buchwald-Hartwig ! Ullman discovers ipso-substitution of aryl halides mediated by copper in 1901 CuX X + HNR2 HNR2 Lindley, J. Tetrahedron, 1984, 40, 1433 ! Scope has been expanded to include a tremendous variety of nucleophiles ! Limited by harsh reaction conditions, stoichiometric metal ! Multiple mechanisms thought to be operating, catalytic species poorly defined ! Aryne chemistry allows for amination of an expanded scope of aryl halides R R R NaNH HNR2 X 2 NR2 Biehl, E. J. Org. Chem., 1987, 52, 2619 ! Functional group compatibility low ! Regiocontrol is a problem The Move Towards a General Reaction ! Bunnett introduces the S RN1 mechanism to the picture Me Me NH3, hv Me Br + Me N - N CH2 Me Me 87% yield Bunnett, J . Acc. Chem. Res. , 1978, 11, 413 ! Demanding couplings can be accomplished ! Reaction conditions are mildest yet ! All drawbacks associated with radical mechanisms are present ! Migita makes the major breakthrough PdCl2(o-tolyl3P)2 Br + Bu3SnNEt2 NEt2 PhCH3, reflux 81% yield Kosugi, M.; Kameyama, M.; Migita, T.