Driving Recursive Dehydration by PIII/PV Catalysis: Annulation of Amines and Carboxylic Acids by Sequential C–N and C–C Bond Formation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UMPOLUNG in REACTIONS CATALYZED by THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung En Reacciones Catalizadas Por Enzimas Dependientes De Pirofosfato De Tiamina
Ciencia, Ambiente y Clima, Vol. 2, No. 2, julio-diciembre, 2019 • ISSN (impreso): 2636-2317 • ISSN (en línea): 2636-2333 DOI: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 UMPOLUNG IN REACTIONS CATALYZED BY THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung en reacciones catalizadas por enzimas dependientes de pirofosfato de tiamina Carlos José Boluda Emily Soto Instituto Tecnológico de Santo Domingo (INTEC), Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales, Av. de Los Área de Ciencias Básicas y Ambientales Próceres 49, Santo Domingo, República Dominicana Correo-e: [email protected] *Corresponding author: Carlos J. Boluda Darah de la Cruz Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Carolina Juncá Área de Ciencias Básicas y Ambientales Instituto Tecnológico de Santo Domingo (INTEC), Correo-e: [email protected] Área de Ciencias Básicas y Ambientales Anny Peña Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales Correo-e: [email protected] Recibido: 25/9/2019 • Aprobado: 19/10/2019 Cómo citar: Boluda, C. J., Juncá, C., Soto, E., de la Cruz, D., & Peña, A. (2019). Umpolung in reactions catalyzed by thiamine pyrophos- phate dependent enzymes. Ciencia, Ambiente Y Clima, 2(2), 27-42. Doi: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 Abstract Resumen The temporal exchange of the electrophilic/nucleophilic El intercambio temporal del carácter electrofílico/nucleofí- character of an atom by chemical manipulation is known lico de un átomo mediante manipulación química, es cono- in organic chemistry as umpolung. This inversion of polarity cido con el vocablo alemán de umpolung. -

Recent Syntheses of Steroidal Oxazoles, Oxazolines and Oxazolidines
A Platinum Open Access Journal Review for Organic Chemistry Free to Authors and Readers DOAJ Seal Arkivoc 2021, part i, 471-490 Recent syntheses of steroidal oxazoles, oxazolines and oxazolidines Besma Bendif,a,b Malika Ibrahim-Ouali,*a and Frédéric Dumur c aAix Marseille Univ, CNRS, Centrale Marseille, iSm2, F-13397 Marseille, France bLaboratoire de Chimie Appliquée, Faculté des Sciences, Université du 08 mai 1945 Guelma, Algeria cAix Marseille Univ, CNRS, ICR, UMR 72 73, F-13397 Marseille, France Email: [email protected] Received 03-15-2021 Accepted 04-11-2021 Published on line 05-08-2021 Abstract It was found that the introduction of heterocycles to steroids often leads in a change of their physiological activity and the appearance of new interesting biological precursors. Recent developments in the syntheses of steroidal oxazoles, oxazolines, and oxazolidines are described herein. The biological activities of those steroidal derivatives for which data are available are given. Keywords: Steroids, oxazoles, oxazolines, oxazolidines DOI: https://doi.org/10.24820/ark.5550190.p011.512 Page 471 ©AUTHOR(S) Arkivoc 2021, i, 471-490 Bendif, B. et al. Table of Contents 1. Introduction 2. Synthesis of Steroidal Oxazoles 3. Synthesis of Steroidal Oxazolines 4. Synthesis of Steroidal Oxazolidines 5. Conclusions Acknowledgements References 1. Introduction Steroids constitute an extensive and important class of biologically active polycyclic compounds that are widely used for therapeutic purposes.1-3 Even after decades of research, the total synthesis of steroid nuclei by improved strategies continues to receive considerable attention. Numerous methods have been exploited for the total synthesis of steroids which are widely distributed in nature and which possess practical medical importance. -
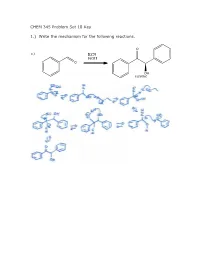
CHEM 345 Problem Set 18 Key 1.) Write the Mechanism for The
CHEM 345 Problem Set 18 Key 1.) Write the mechanism for the following reactions. O a.) KCN EtOH O OH racemic 1.) Write the mechanism for the following reactions. b.) KCN AcOH O NC OH racemic O c.) N S R O NEt3 OH racemic 2.) What is the structure of AcOH?Why does changing the solvent from EtOH to AcOH make such a big difference? O OH AcOH acetic acid The pKa of acetic acid is approximately 5. The pKa of ethanol is approximately 15. When you take a proton off of ethanol, you generate ethoxide which is about 1010 times stronger of a base than acetate. 3.) Give two instances when you need to use the thiazolium salt and triethylamine rather than KCN and EtOH. If the aldehydes contain an enolizable proton then you cannot use KCN/EtOH, instead you must use the thiazolium. Also, if the electrophile is a Michael acceptor to give a 1,4 dicarbonyl, then the thiazolium catalyst should be used. 4.) Break the following compound down as far as you can using Aldol, Michael, and Claisen reactions. Above each retrosynthetic arrow, write the name of the reaction. O HO O HO Aldol O Michael HO O O HO O Aldol O O Aldol HO O O HO Michael O O O Aldol There are other possibilities for order. O O HO Aldol O O 5.) Synthesize the following molecules. All carbons in the molecules must come from benzene or compounds with 5C’s or less. a.) O H2SO4 O HNO3 O2N AlCl3 O O SOCl2 HO Cl H2CrO4 1.) BuLi, Et2O + O 2.) H3O HO b.) O O O Cl + H3O NaOEt, EtOH O O O O Cl O AlCl3 Cl Cl AlCl3 Cl2 c.) O OMe NaOMe MeOH O O 1.) POCl3, DMF 2.) H2O OMe OMe O AlCl3 MeI Cl ONa HCl NaOH ZnHg 1.) NaOH + mcpba O 2.) H3O O OH O O AlCl3 Cl 6.) Write the mechanism for the following reactions. -

DEVELOPMENT of NOVEL METHODOLOGIES UTILIZING QUATERNARY AMMONIUM SALTS AS CATALYSTS by LINDSEY RAE CULLEN DISSERTATION Submitted
DEVELOPMENT OF NOVEL METHODOLOGIES UTILIZING QUATERNARY AMMONIUM SALTS AS CATALYSTS BY LINDSEY RAE CULLEN DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate College of the University of Illinois at Urbana-Champaign, 2015 Urbana, Illinois Doctoral Committee: Professor Scott E. Denmark, Chair Professor Martin D. Burke Professor Scott K. Silverman Professor Steven C. Zimmerman ii Abstract The first half of this thesis (Chapter 2) described the development of a fluoride-promoted conjugate addition of sulfur-stabilized carbanion nucleophiles to α,β-unsaturated ketones and esters. This reaction was achieved using a substoichiometric amount of TBAF, resulting in high yields on the desired 1,4-addition product. The addition of 1,3-dithianes was given particular focus as a novel method for the preparation of differentially protect 1,4-dicarbonyl compounds. Observation by 13C NMR spectroscopy provided evidence that the reaction proceeds through an ion pair, and attempts to extend this reaction to asymmetric additions using a chiral counterion are presented in detail. The second half of this thesis (Chapter 3) details development of a phase transfer catalyzed [2,3]-sigmatropic rearrangement of allyloxy carbonyl compounds. Initial investigation focused on identifying viable substrate classes that would undergo selective [2,3]-rearrangement under phase transfer conditions. Under certain conditions, the [2,3]-sigmatropic rearrangement of allyloxy carbonyl compounds takes place in the presence of a phase transfer agent, providing a rare example of a phase transfer catalyzed unimolecular reaction. In the course of this investigation it was found that catalysis is dependent on several variables including base concentration, catalyst structure, and substrate lipophilicity. -

Nickel-Catalyzed Cyanation of Benzylic and Allylic Pivalates
Nickel-Catalyzed Cyanation of Benzylic and Allylic Pivalates by Alexandria Daria Maria Jeanneret A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Chemistry University of Toronto © Copyright by Alexandria Daria Maria Jeanneret 2018 Nickel-Catalyzed Cyanation of Benzylic and Allylic Pivalates Alexandria Daria Maria Jeanneret Master of Science Department of Chemistry University of Toronto 2018 Abstract Nitriles are considered very versatile functional groups due to their ability to easily be transformed into a variety of other functional groups in one or two steps. In particular, the synthesis of α-arylnitriles is of interest to organic chemists due to their presence in pharmaceuticals and their value as synthetic intermediates. Taking advantage of nickel’s unique ability to insert into a C‒O bond, the focus of this thesis is on the nickel-catalyzed cyanation of benzylic and allylic pivalates, exploring the use of inorganic and organic cyanide sources for this transformation. The substrate scope for the synthesis of benzylic and allylic nitriles will be presented as well as studies examining the functional group tolerance of this cyanation reaction, which led to further insights into the mechanism and applicability of this chemistry. ii Acknowledgments First and foremost, I would like to thank Professor Sophie Rousseaux for the opportunity to work to work in her lab over the last year. Her passion for chemistry is beyond contagious and her guidance has been invaluable. I would also like to thank Professor Mark Taylor for his help with this thesis. Secondly, I would like to thank all the staff at the NMR and AIMS facility for all their hard-work and dedication to ensuring the instruments run smoothly and for always taking the time to answer questions. -

Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— a Literature Review
Molecules 2013, 18, 11873-11903; doi:10.3390/molecules181011873 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— A Literature Review Sowmini Kumaran 1, Mulchand S. Patel 2 and Frank Jordan 1,* 1 Department of Chemistry, Rutgers University, Newark, NJ 07102, USA 2 Department of Biochemistry, School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14214, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-973-353-5470; Fax: +1-973-353-1264. Received: 30 July 2013; in revised form: 14 September 2013 / Accepted: 16 September 2013 / Published: 26 September 2013 Abstract: The 2-oxoacid dehydrogenase complexes (ODHc) consist of multiple copies of three enzyme components: E1, a 2-oxoacid decarboxylase; E2, dihydrolipoyl acyl-transferase; and E3, dihydrolipoyl dehydrogenase, that together catalyze the oxidative decarboxylation of 2-oxoacids, in the presence of thiamin diphosphate (ThDP), coenzyme A 2+ + (CoA), Mg and NAD , to generate CO2, NADH and the corresponding acyl-CoA. The structural scaffold of the complex is provided by E2, with E1 and E3 bound around the periphery. The three principal members of the family are pyruvate dehydrogenase (PDHc), 2-oxoglutarate dehydrogenase (OGDHc) and branched-chain 2-oxo acid dehydrogenase (BCKDHc). In this review, we report application of NMR-based approaches to both mechanistic and structural issues concerning these complexes. These studies revealed the nature and reactivity of transient intermediates on the enzymatic pathway and provided site-specific information on the architecture and binding specificity of the domain interfaces using solubilized truncated domain constructs of the multi-domain E2 component in its interactions with the E1 and E3 components. -

Regioselective Synthesis of Enol Triflates by Enolate Trapping
Reviews and Accounts ARKIVOC 2012 (i) 432-490 The Hendrickson ‘POP’ reagent and analogues thereof: synthesis, structure, and application in organic synthesis Ziad Moussa Department of Chemistry, Taibah University, P.O. Box 30002, Almadinah Almunawarrah, Saudi Arabia E-mail: [email protected] DOI: http://dx.doi.org/10.3998/ark.5550190.0013.111 Abstract This comprehensive review discusses the synthesis, structure, and application of the Hendrickson ‘POP’ reagent and its analogues in organic synthesis. Specifically, the review describes applications of the preceding dehydrating agents in functional group interconversions, heterocycle synthesis, and total synthesis of natural products. Many examples are provided from the literature to show the scope and selectivity (regio and chemo) in these transformations. Keywords: Hendrickson ‘POP’ reagent, functional group interconversions, heterocycle synthesis, total synthesis of natural products Table of Contents 1. Introduction 2. The Hendrickson Reagent 2.1. Preparation of the Hendrickson reagent 2.2. Application of the Hendrickson reagent in functional group interconversions 2.2.1. Conversion of aldoximes into nitriles 2.2.2. Conversion of carboxylic acids to benzimidazole, benzoxazole, and benzthiazole heterocycles 2.2.3. Amide and amidine synthesis 2.2.4. Conversion of carboxylic acids into acid anhydrides 2.2.5. Ester formation from acid and alcohol 2.2.6. Conversion of activated ketones to alkynes 2.2.7. Cyclodehydrations of diols and amino alcohols 2.2.8. Conversion of epoxides to dienes 2.2.8.1 Mechanism of double elimination of epoxides to dienes Page 432 ©ARKAT-USA, Inc. Reviews and Accounts ARKIVOC 2012 (i) 432-490 2.2.9. Conversion of sulfonic acids to sulfonamides and activated sulfonate esters 2.2.10. -

A Biosynthetically Inspired Synthetic Route to Substituted Furans, and Its Application to the Total Synthesis of the Furan Fatty
A Biosynthetically Inspired Synthetic Route to Substituted Furans, and its Application to the Total Synthesis of the Furan Fatty Acid F5 A thesis submitted in partial fulfilment of the requirements of the degree Doctor of Philosophy Robert Jack Lee Supervisors: Dr Gareth J. Pritchard and Dr Marc C. Kimber 1 Thesis Abstract Dietary fish oil supplementation has long been shown to have significant health benefits, largely stemming from the anti-inflammatory activity of the ω-3 and ω-6 polyunsaturated fatty acids (PUFAs) present in fish oils. The anti-inflammatory properties of these fatty acids has been linked to beneficial health effects, such as protecting the heart, in individuals consuming diets rich in fish, or supplemented with fish oils.1 These effects are highly notable in the Māori people native to coastal regions of New Zealand; the significantly lower rates of heart problems compared to the inland populous has been attributed to the consumption of the green lipped mussel Perna Canaliculus. Commercially available health supplements based on the New Zealand green lipped mussel include a freeze-dried powder and a lipid extract (Lyprinol®), the latter of which has shown anti- inflammatory properties comparable to classical non-steroidal anti-inflammatory drugs (NSAIDs) such as Naproxen.2 GCMS analysis of Lyprinol by Murphy et al. showed the presence of a class of ω-4 and ω-6 PUFAs bearing a highly electron rich tri- or tetra-alkyl furan ring, which were designated furan fatty acids (F-acids).3 Due to their instability, isolation of F- acids from natural sources cannot be carried out and a general synthetic route toward this class of natural products was required. -

Activation of Alcohols Toward Nucleophilic Substitution: Conversion of Alcohols to Alkyl Halides Amani Atiyalla Abdugadar
University of Northern Colorado Scholarship & Creative Works @ Digital UNC Theses Student Research 12-1-2012 Activation of Alcohols Toward Nucleophilic Substitution: Conversion of Alcohols to Alkyl Halides Amani Atiyalla Abdugadar Follow this and additional works at: http://digscholarship.unco.edu/theses Recommended Citation Abdugadar, Amani Atiyalla, "Activation of Alcohols Toward Nucleophilic Substitution: Conversion of Alcohols to Alkyl Halides" (2012). Theses. Paper 22. This Text is brought to you for free and open access by the Student Research at Scholarship & Creative Works @ Digital UNC. It has been accepted for inclusion in Theses by an authorized administrator of Scholarship & Creative Works @ Digital UNC. For more information, please contact [email protected]. © 2012 Amani Abdugadar ALL RIGHTS RESERVED UNIVERSITY OF NORTHERN COLORADO Greeley, Colorado The Graduate School ACTIVATION OF ALCOHOLS TOWARD NEOCLEOPHILIC SUBSTITUTION: CONVERSION OF ALCOHOLS TO ALKYL HALIDES A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science Amani Abdugadar College of Natural and Health Sciences Department of Chemistry and Biochemistry December, 2012 This Thesis by: Amani Abdugadar Entitled: Activation of Alcohols Toward Neocleophilic Substitution: Conversion of Alcohols to Alkyl Halides has been approved as meeting the requirement for the Master of Science in College of Natural and Health Sciences in Department of Chemistry and Biochemistry Accepted by the Thesis Committee ______________________________________________________ Michael D. Mosher, Ph.D., Research Co-Advisor ______________________________________________________ Richard W. Schwenz, Ph.D., Research Co-Advisor ______________________________________________________ David L. Pringle, Ph.D., Committee Member Accepted by the Graduate School _________________________________________________________ Linda L. Black, Ed.D., LPC Acting Dean of the Graduate School and International Admissions ABSTRACT Abdugadar, Amani. -

Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids
International Journal of Molecular Sciences Article Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids Stefan R. Marsden, Duncan G. G. McMillan and Ulf Hanefeld * Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Van der Maasweg 9, 2629HZ Delft, The Netherlands; [email protected] (S.R.M.); [email protected] (D.G.G.M.) * Correspondence: [email protected] Received: 12 October 2020; Accepted: 12 November 2020; Published: 16 November 2020 Abstract: The synthetic properties of the Thiamine diphosphate (ThDP)-dependent pyruvate dehydrogenase E1 subunit from Escherichia coli (EcPDH E1) was assessed for carboligation reactions with aliphatic ketoacids. Due to its role in metabolism, EcPDH E1 was previously characterised with respect to its biochemical properties, but it was never applied for synthetic purposes. Here, we show that EcPDH E1 is a promising biocatalyst for the production of chiral α-hydroxyketones. WT EcPDH E1 shows a 180–250-fold higher catalytic efficiency towards 2-oxobutyrate or pyruvate, respectively, in comparison to engineered transketolase variants from Geobacillus stearothermophilus (TKGST). Its broad active site cleft allows for the efficient conversion of both (R)- and (S)-configured α-hydroxyaldehydes, next to linear and branched aliphatic aldehydes as acceptor substrates under kinetically controlled conditions. The alternate, thermodynamically controlled self-reaction of aliphatic aldehydes was shown to be limited to low levels of conversion, which we propose to be due to their large hydration constants. Additionally, the thermodynamically controlled approach was demonstrated to suffer from a loss of stereoselectivity, which makes it unfeasible for aliphatic substrates. Keywords: Thiamine diphosphate; transketolase; C-C bond formation; kinetic control; acyloins 1. -
![Umpolung of Amine Reactivity. Nucleophilic [Alpha]](https://docslib.b-cdn.net/cover/9062/umpolung-of-amine-reactivity-nucleophilic-alpha-2689062.webp)
Umpolung of Amine Reactivity. Nucleophilic [Alpha]
11591 S. W. Benson: Thermochemical Kinetics. Wiley, New York, N. Y., [I631 C. M. Shy and J. F. Finklea, Environ. Sci. Technol. 7, 205 (1973). 1968. [I641 R. P. Steer, K. R. Darnall, and J. N. Pifrs, Jr., Tetrahedron Lett. [I601 J. G. Caluert, K. L. Demerjian, and J. A. Kerr, Proc. Int. Symp. 1969. 3765. Air Pollut., Tokyo, Oct. 17-19, 1972, pp. 465ff. 11651 S. Furuyama. R. Atkinson, A. J. Colussi, and R. J. Cvetanouic, Int. [161] A. Q. Eschenroeder and J. R. Marfinez, Advan. Chem. Ser. 113, 101 J. Chem. Kinet. 6, 741 (1974). (1972). [166] J. N. Pitrs, Jr., P. G. Bekowies. G. J. Doyle, J. M. McAfee, and [I621 7: A. Hecht, J. H. Seinfeld. and M. C. Dodge, Environ. Sci. Technol. A. M. Winer, to be oublished. 8, 327 (1974). and references therein. Umpolung of Amine Reactivity. Nucleophilic a-(Secondary Amino)- New synthetic alky lation via Metalated Nitrosaminesr**lr”’l methods 0 By Dieter Seebach and Dieter Ended*] There are basically two kinds of hetero atoms in organic molecules: one kind confers electrophilic character upon the carbon atom to which it is bound, and the other kind turns it into a nucleophilic site. The development of methods permitting transitions between the two resulting categories of reagents has become an important task of modern organic synthesis. The scope of such umpolung of the reactivity of functional groups is discussed for the case of amines as an example. A method of preparing masked u-secondary amino carbanions consists in nitrosation of the secondary amine, followed by metalation of the resulting nitrosamine CL to the nitrogen, reaction with electrophiles, and subsequent denitrosation. -

Organic Chemistry 2 Ester Enolates and Acyl Anion Equivalents
Dr. Peter Wipf Chemistry 0320 - Organic Chemistry 2 Ester Enolates and Acyl Anion Equivalents The Claisen condensation is the ester analog of the aldol reaction. Under standard conditions (sodium alkoxide, alkohol), the equilibrium is shifted to the β-dicarbonyl compound as a consequence of the irreversible enolization to the highly resonance-stabilized dicarbonyl enolate. O O NaOR, HOR O O O O + OR R' R' R' R'=OR, C The intramolecular Claisen reaction is generally called the Dieckmann condensation. Another modification of the Claisen condensation is the use of strong base which leads to irreversible enolization of the carbonyl compounds. O OLi LDA, -78 °C PhCOCl O O OEt OEt THF Ph OEt β-Dicarbonyl, and, generally, active methylene compounds provide a rich source of chemistry in alkylation reactions, due to their relative ease of enolization and the possibility for removal of one of the activating groups. Z1 Z2 , Z = ester, ketone, amide, nitrile, aldehyde, nitro, sulfoxide, sulfone, sulfonate, sulfonamide H H Especially important are acetoacetate and malonate building blocks (acetoacetic and malonic ester syntheses): O O O O O O OEt OEt OEt XCH2CO2R R-X R-X (1 equiv) RCOX O O O O OH O O R R OEt O R Dithioketals can be used in Umpolung reactions as latent carbonyl groups. Nature’s Umpolung reagent is thiamine pyrophosphate (TPP). In the pentose phosphate pathway, TPP catalyzes the conversion of erythrose into fructose. First, the carbanion of TPP attacks the carbonyl carbon of xylulose 5-phosphate, forming a proton from the TPP adduct, and subsequent electron rearrangement leads to fragmentation, releasing the first product, glyceraldehyde 3-phosphate.