Development of Pyruvate:Ferredoxin Oxidoreductase Inhibitors for the Treatment of Clostridium Difficile Infection
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UMPOLUNG in REACTIONS CATALYZED by THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung En Reacciones Catalizadas Por Enzimas Dependientes De Pirofosfato De Tiamina
Ciencia, Ambiente y Clima, Vol. 2, No. 2, julio-diciembre, 2019 • ISSN (impreso): 2636-2317 • ISSN (en línea): 2636-2333 DOI: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 UMPOLUNG IN REACTIONS CATALYZED BY THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung en reacciones catalizadas por enzimas dependientes de pirofosfato de tiamina Carlos José Boluda Emily Soto Instituto Tecnológico de Santo Domingo (INTEC), Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales, Av. de Los Área de Ciencias Básicas y Ambientales Próceres 49, Santo Domingo, República Dominicana Correo-e: [email protected] *Corresponding author: Carlos J. Boluda Darah de la Cruz Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Carolina Juncá Área de Ciencias Básicas y Ambientales Instituto Tecnológico de Santo Domingo (INTEC), Correo-e: [email protected] Área de Ciencias Básicas y Ambientales Anny Peña Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales Correo-e: [email protected] Recibido: 25/9/2019 • Aprobado: 19/10/2019 Cómo citar: Boluda, C. J., Juncá, C., Soto, E., de la Cruz, D., & Peña, A. (2019). Umpolung in reactions catalyzed by thiamine pyrophos- phate dependent enzymes. Ciencia, Ambiente Y Clima, 2(2), 27-42. Doi: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 Abstract Resumen The temporal exchange of the electrophilic/nucleophilic El intercambio temporal del carácter electrofílico/nucleofí- character of an atom by chemical manipulation is known lico de un átomo mediante manipulación química, es cono- in organic chemistry as umpolung. This inversion of polarity cido con el vocablo alemán de umpolung. -
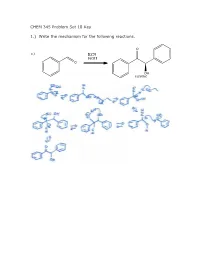
CHEM 345 Problem Set 18 Key 1.) Write the Mechanism for The
CHEM 345 Problem Set 18 Key 1.) Write the mechanism for the following reactions. O a.) KCN EtOH O OH racemic 1.) Write the mechanism for the following reactions. b.) KCN AcOH O NC OH racemic O c.) N S R O NEt3 OH racemic 2.) What is the structure of AcOH?Why does changing the solvent from EtOH to AcOH make such a big difference? O OH AcOH acetic acid The pKa of acetic acid is approximately 5. The pKa of ethanol is approximately 15. When you take a proton off of ethanol, you generate ethoxide which is about 1010 times stronger of a base than acetate. 3.) Give two instances when you need to use the thiazolium salt and triethylamine rather than KCN and EtOH. If the aldehydes contain an enolizable proton then you cannot use KCN/EtOH, instead you must use the thiazolium. Also, if the electrophile is a Michael acceptor to give a 1,4 dicarbonyl, then the thiazolium catalyst should be used. 4.) Break the following compound down as far as you can using Aldol, Michael, and Claisen reactions. Above each retrosynthetic arrow, write the name of the reaction. O HO O HO Aldol O Michael HO O O HO O Aldol O O Aldol HO O O HO Michael O O O Aldol There are other possibilities for order. O O HO Aldol O O 5.) Synthesize the following molecules. All carbons in the molecules must come from benzene or compounds with 5C’s or less. a.) O H2SO4 O HNO3 O2N AlCl3 O O SOCl2 HO Cl H2CrO4 1.) BuLi, Et2O + O 2.) H3O HO b.) O O O Cl + H3O NaOEt, EtOH O O O O Cl O AlCl3 Cl Cl AlCl3 Cl2 c.) O OMe NaOMe MeOH O O 1.) POCl3, DMF 2.) H2O OMe OMe O AlCl3 MeI Cl ONa HCl NaOH ZnHg 1.) NaOH + mcpba O 2.) H3O O OH O O AlCl3 Cl 6.) Write the mechanism for the following reactions. -

Structural Insights Into the Mechanism of Inhibition of AHAS by Herbicides
Structural insights into the mechanism of inhibition of PNAS PLUS AHAS by herbicides Thierry Lonhiennea,1,2, Mario D. Garciaa,1, Gregory Pierensb, Mehdi Moblia,b, Amanda Nouwensa, and Luke W. Guddata,2 aSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, QLD 4072, Australia; and bCenter for Advanced Imaging, The University of Queensland, Brisbane, QLD 4072, Australia Edited by María-Jazmin Abraham-Juarez, University of California, Berkeley, CA, and accepted by Editorial Board Member Gregory A. Petsko January 22, 2018 (received for review August 16, 2017) Acetohydroxyacid synthase (AHAS), the first enzyme in the FAD cofactor is required for the enzyme to be active (14). The branched amino acid biosynthesis pathway, is present only in reversible accumulative inhibition described here is not equivalent plants and microorganisms, and it is the target of >50 commercial to the “reversible time-dependent inhibition” described by herbicides. Penoxsulam (PS), which is a highly effective broad- Morrison and Walsh (15). Reversible time-dependent inhibition spectrum AHAS-inhibiting herbicide, is used extensively to control generally describes a process in which the inhibitor–enzyme weed growth in rice crops. However, the molecular basis for its complex requires time to acquire a conformation in which the inhibition of AHAS is poorly understood. This is despite the avail- inhibitor is fully effective. In that mode of inhibition, reversibility ability of structural data for all other classes of AHAS-inhibiting herbicides. Here, crystallographic data for Saccharomyces cerevisiae relates to the binding alone, with the inhibitor being able to AHAS (2.3 Å) and Arabidopsis thaliana AHAS (2.5 Å) in complex dissociate from the complex. -

Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— a Literature Review
Molecules 2013, 18, 11873-11903; doi:10.3390/molecules181011873 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— A Literature Review Sowmini Kumaran 1, Mulchand S. Patel 2 and Frank Jordan 1,* 1 Department of Chemistry, Rutgers University, Newark, NJ 07102, USA 2 Department of Biochemistry, School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14214, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-973-353-5470; Fax: +1-973-353-1264. Received: 30 July 2013; in revised form: 14 September 2013 / Accepted: 16 September 2013 / Published: 26 September 2013 Abstract: The 2-oxoacid dehydrogenase complexes (ODHc) consist of multiple copies of three enzyme components: E1, a 2-oxoacid decarboxylase; E2, dihydrolipoyl acyl-transferase; and E3, dihydrolipoyl dehydrogenase, that together catalyze the oxidative decarboxylation of 2-oxoacids, in the presence of thiamin diphosphate (ThDP), coenzyme A 2+ + (CoA), Mg and NAD , to generate CO2, NADH and the corresponding acyl-CoA. The structural scaffold of the complex is provided by E2, with E1 and E3 bound around the periphery. The three principal members of the family are pyruvate dehydrogenase (PDHc), 2-oxoglutarate dehydrogenase (OGDHc) and branched-chain 2-oxo acid dehydrogenase (BCKDHc). In this review, we report application of NMR-based approaches to both mechanistic and structural issues concerning these complexes. These studies revealed the nature and reactivity of transient intermediates on the enzymatic pathway and provided site-specific information on the architecture and binding specificity of the domain interfaces using solubilized truncated domain constructs of the multi-domain E2 component in its interactions with the E1 and E3 components. -

An Escherichiacoli Mutant Deficient in Pyruvate Oxidase Activity Due To
Proc. Natl. Acad. Sci. USA Vol. 81, pp. 4348-4352, July 1984 Biochemistry An Escherichia coli mutant deficient in pyruvate oxidase activity due to altered phospholipid activation of the enzyme (lipid activation/lipid binding/hydrophobic site/conformational change/proteolytic activation) YING-YING CHANG AND JOHN E. CRONAN, JR. Department of Microbiology, University of Illinois, 131 Burrill Hall, 407 South Goodwin, Urbana, IL 61801 Communicated by Ralph S. Wolfe, April 4, 1984 ABSTRACT The pyruvate oxidase (pyruvate:ferricyto- lipid activation of an enzyme occurs in vivo and is physiolog- chrome b, oxidoreductase, EC 1.2.2.2) of Escherichia coli is ically significant. markedly activated by phospholipids in vitro. To test the phys- iological relevance of this activation, we isolated an E. coil mu- MATERIALS AND METHODS tant producing an oxidase that is deficient in activation by (and binding to) phospholipids. The mutant oxidase could be Bacterial Strains, Media, and Extract Preparation. Strains fully activated by a specific proteolytic cleavage, indicating CY265 (pox') and YYC124 (poxB3) are isogenic derivatives that the catalytic site is normal. The mutant enzyme functions (4) of E. coli K-12 that carry a deletion of the aceEF (pyru- poorly in vivo, indicating that activation of the oxidase by vate dehydrogenase) genes. Strain YYC124 is a pyruvate ox- phospholipids plays an important physiological role. idase mutant derived from strain CY265 (4). The strains were grown in broth containing 10 mM sodium acetate at 330C, Enzyme activation by membrane phospholipids in vitro has and cell extracts were prepared as described (3, 4). In some often been observed (1, 2) and is generally ascribed a physio- cases, the cell-free extract was heated to 600C for 5 min and logical role. -

Recent Progress in the Microbial Production of Pyruvic Acid
fermentation Review Recent Progress in the Microbial Production of Pyruvic Acid Neda Maleki 1 and Mark A. Eiteman 2,* 1 Department of Food Science, Engineering and Technology, University of Tehran, Karaj 31587-77871, Iran; [email protected] 2 School of Chemical, Materials and Biomedical Engineering, University of Georgia, Athens, GA 30602, USA * Correspondence: [email protected]; Tel.: +1-706-542-0833 Academic Editor: Gunnar Lidén Received: 10 January 2017; Accepted: 6 February 2017; Published: 13 February 2017 Abstract: Pyruvic acid (pyruvate) is a cellular metabolite found at the biochemical junction of glycolysis and the tricarboxylic acid cycle. Pyruvate is used in food, cosmetics, pharmaceutical and agricultural applications. Microbial production of pyruvate from either yeast or bacteria relies on restricting the natural catabolism of pyruvate, while also limiting the accumulation of the numerous potential by-products. In this review we describe research to improve pyruvate formation which has targeted both strain development and process development. Strain development requires an understanding of carbohydrate metabolism and the many competing enzymes which use pyruvate as a substrate, and it often combines classical mutation/isolation approaches with modern metabolic engineering strategies. Process development requires an understanding of operational modes and their differing effects on microbial growth and product formation. Keywords: auxotrophy; Candida glabrata; Escherichia coli; fed-batch; metabolic engineering; pyruvate; pyruvate dehydrogenase 1. Introduction Pyruvic acid (pyruvate at neutral pH) is a three carbon oxo-monocarboxylic acid, also known as 2-oxopropanoic acid, 2-ketopropionic acid or acetylformic acid. Pyruvate is biochemically located at the end of glycolysis and entry into the tricarboxylic acid (TCA) cycle (Figure1). -

United States Patent (19) 11 Patent Number: 4,458,686 Clark, Jr
United States Patent (19) 11 Patent Number: 4,458,686 Clark, Jr. 45) Date of Patent: Jul. 10, 1984 (54), CUTANEOUS METHODS OF MEASURING 4,269,516 5/1981 Lubbers et al. ..................... 356/427 BODY SUBSTANCES 4,306,877 12/1981 Lubbers .............................. 128/633 Primary Examiner-Benjamin R. Padgett 75) Inventor: Leland C. Clark, Jr., Cincinnati, Ohio Assistant Examiner-T. J. Wallen 73. Assignee: Children's Hospital Medical Center, Attorney, Agent, or Firm-Wood, Herron & Evans Cincinnati, Ohio 57 ABSTRACT (21) Appl. No.: 491,402 Cutaneous methods for measurement of substrates in 22 Filed: May 4, 1983 mammalian subjects are disclosed. A condition of the skin is used to measure a number of important sub - Related U.S. Application Data stances which diffuse through the skin or are present (62) Division of Ser. No. 63,159, Aug. 2, 1979, Pat. No. underneath the skin in the blood or tissue. According to 4,401,122. the technique, an enzyme whose activity is specific for a particular substance or substrate is placed on, in or 51). Int. Cl. ................................................ A61B5/00 under the skin for reaction. The condition of the skin is 52). U.S. Cl. .................................... 128/635; 128/636; then detected by suitable means as a measure of the 436/11 amount of the substrate in the body. For instance, the (58), Field of Search ............... 128/632, 635, 636, 637, enzymatic reaction or by-product of the reaction is 128/633; 356/41, 417, 427; 422/68; 23/230 B; detected directly through the skin as a measure of the 436/11 amount of substrate. -

Rapid Expression and Purification of 100 Nmol Quantities of Active Protein Using Cell-Free Protein Synthesis
102 Biotechnol. Prog. 2004, 20, 102−109 Rapid Expression and Purification of 100 nmol Quantities of Active Protein Using Cell-Free Protein Synthesis Michael C. Jewett and James R. Swartz* Department of Chemical Engineering, Stanford University, Stanford, California 94305-5025 Two strategies for ATP regeneration during cell-free protein synthesis were applied to the large-scale production and single-column purification of active chloramphenicol acetyl transferase (CAT). Fed-batch reactions were performed on a 5-10 mL scale, approximately 2 orders of magnitude greater than the typical reaction volume. The pyruvate oxidase system produced 104 nmol of active CAT ina5mLreaction over the course of 5 h. The PANOx system produced 261 ( 42 nmol, about 7 mg, of active CAT in a 10 mL reaction over the course of 4 h. The reaction product was purified to apparent homogeneity with approximately 70% yield by a simple affinity chromatog- raphy adsorption and elution. To our knowledge, this is the largest amount of actively expressed protein to be reported in a simple, fed-batch cell-free protein synthesis reaction. Introduction enous acetate kinase present in the cell extract prepared from E. coli. The most notable feature of this system is Cell-free protein synthesis offers a method to exploit that phosphate released during protein synthesis is biological processes without intact cells. It is a convenient recycled into acetyl-phosphate (Figure 1A). This attribute tool for the production of active recombinant DNA (rDNA) allows for numerous additions of the energy source proteins and has several advantages over in vivo protein without inhibition of the protein synthesis reaction via expression systems (1-4). -

Oxoglutarate Dehydrogenase Multienzyme Complexes Reconstituted from Recombinant
*RevisedView metadata, Manuscript citation and (text similar UNmarked) papers at core.ac.uk brought to you by CORE Click here to view linked References provided by Repository of the Academy's Library Formation of reactive oxygen species by human and bacterial pyruvate and 2- oxoglutarate dehydrogenase multienzyme complexes reconstituted from recombinant components Attila Ambrusa, Natalia S. Nemeriab, Beata Torocsika, Laszlo Trettera, Mattias Nilssona, Frank Jordanb, and Vera Adam-Vizia,* aDepartment of Medical Biochemistry, MTA-SE Laboratory for Neurobiochemistry, Semmelweis University, Budapest, 1094, Hungary bDepartment of Chemistry, Rutgers, the State University, Newark, NJ 07102, USA *To whom correspondence should be addressed at: Department of Medical Biochemistry, Semmelweis University, 37-47 Tuzolto Street, Budapest, 1094, Hungary. Tel.: +361 266 2773, Fax.: +361 267 0031, E-mail: [email protected] 1 Abstract Individual recombinant components of pyruvate and 2-oxoglutarate dehydrogenase multienzyme complexes (PDHc, OGDHc) of human and Escherichia coli (E. coli) origin were expressed and purified from E. coli with optimized protocols. The four multienzyme complexes were each reconstituted under optimal conditions at different stoichiometric ratios. Binding stoichiometries for the highest catalytic efficiency were determined from the rate of NADH generation by the complexes at physiological pH. Since some of these complexes were shown to possess ‘moonlighting’ activities under pathological conditions often accompanied by acidosis, activities were also determined at pH 6.3. As reactive oxygen species (ROS) generation by the E3 component of hOGDHc is a pathologically relevant feature, superoxide generation by the complexes with optimal stoichiometry was measured by the acetylated cytochrome c reduction method in both the forward and the reverse catalytic directions. -

Pyruvate Oxidase, Its Preparation and Use
ft-: .pr Europaisches Patentamt J European Patent Office ® Publication number: 0 274 4SS Office europeen des brevets A2 EUROPEAN PATENT APPLICATION @ Application number: 68300073.9 Int.CI.*: C 12 N 15/00 C 12 N 9/02, 0 12 Q 1/50, @ Date of filing: 06.01.88 C 12 Q 1/48, C 12 Q 1/26 Priority: 06.01.87 JP 903/87 Imamura, Shlgeyuki 15.05.87 JP 118161/87 696, Mifuku Ohito-cho Tagata-gun Shizuoka-ken (JP) Date of publication of application: 13.07.88 Bulletin 88/28 Sagai, Hitoshi 1 28-51 , Naka MiShima-shi Shizuoka-ken (JP) Designated Contracting States: DE FR IT Misaki, Hidfeo Applicant: TOYO JOZO KABUSHIKI KAISHA 774,Yosh!daOhlto-cho 632-1 Mifuku Ohito-cho Tagata-gun Shizuoka-ken (JP) tagata-gun Shizuoka-ken (JP) Nogata, Keiko 855, Yoka-machi Nirayama-cho Inventor: Matsumura, Eiji Tagata-gun Shizuoka-ken (JP) 530-1, Yoka-machi Nirayama-cho Tagate-gun Shizuoka-ken (JP) Representative : Woods, Geoffrey Corlett et at J.A. KEMP & CO. 14 South Square Gray's Inn London WC1R5EU (GB) @ Pyruvate oxidase, its preparation and use. @ A pyruvate oxidase having the ability to catalyse a reaction from pyruvate, phosphate and oxygen with thS formation of acetylphosphate, carbon dioxide and hydrogen peroxide, an ATP-ass content of below 0.0005% and substantially no lactate oxidase activity is prepared by genetic engineering techniques. A pyruvate oxidase gene is isolated from a known enzymatic source of the enzyme and used to transform a microorganism and provide a transformant which can be cultured to provide substantially pure pyruvate oxidase. -

Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids
International Journal of Molecular Sciences Article Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids Stefan R. Marsden, Duncan G. G. McMillan and Ulf Hanefeld * Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Van der Maasweg 9, 2629HZ Delft, The Netherlands; [email protected] (S.R.M.); [email protected] (D.G.G.M.) * Correspondence: [email protected] Received: 12 October 2020; Accepted: 12 November 2020; Published: 16 November 2020 Abstract: The synthetic properties of the Thiamine diphosphate (ThDP)-dependent pyruvate dehydrogenase E1 subunit from Escherichia coli (EcPDH E1) was assessed for carboligation reactions with aliphatic ketoacids. Due to its role in metabolism, EcPDH E1 was previously characterised with respect to its biochemical properties, but it was never applied for synthetic purposes. Here, we show that EcPDH E1 is a promising biocatalyst for the production of chiral α-hydroxyketones. WT EcPDH E1 shows a 180–250-fold higher catalytic efficiency towards 2-oxobutyrate or pyruvate, respectively, in comparison to engineered transketolase variants from Geobacillus stearothermophilus (TKGST). Its broad active site cleft allows for the efficient conversion of both (R)- and (S)-configured α-hydroxyaldehydes, next to linear and branched aliphatic aldehydes as acceptor substrates under kinetically controlled conditions. The alternate, thermodynamically controlled self-reaction of aliphatic aldehydes was shown to be limited to low levels of conversion, which we propose to be due to their large hydration constants. Additionally, the thermodynamically controlled approach was demonstrated to suffer from a loss of stereoselectivity, which makes it unfeasible for aliphatic substrates. Keywords: Thiamine diphosphate; transketolase; C-C bond formation; kinetic control; acyloins 1. -
![Umpolung of Amine Reactivity. Nucleophilic [Alpha]](https://docslib.b-cdn.net/cover/9062/umpolung-of-amine-reactivity-nucleophilic-alpha-2689062.webp)
Umpolung of Amine Reactivity. Nucleophilic [Alpha]
11591 S. W. Benson: Thermochemical Kinetics. Wiley, New York, N. Y., [I631 C. M. Shy and J. F. Finklea, Environ. Sci. Technol. 7, 205 (1973). 1968. [I641 R. P. Steer, K. R. Darnall, and J. N. Pifrs, Jr., Tetrahedron Lett. [I601 J. G. Caluert, K. L. Demerjian, and J. A. Kerr, Proc. Int. Symp. 1969. 3765. Air Pollut., Tokyo, Oct. 17-19, 1972, pp. 465ff. 11651 S. Furuyama. R. Atkinson, A. J. Colussi, and R. J. Cvetanouic, Int. [161] A. Q. Eschenroeder and J. R. Marfinez, Advan. Chem. Ser. 113, 101 J. Chem. Kinet. 6, 741 (1974). (1972). [166] J. N. Pitrs, Jr., P. G. Bekowies. G. J. Doyle, J. M. McAfee, and [I621 7: A. Hecht, J. H. Seinfeld. and M. C. Dodge, Environ. Sci. Technol. A. M. Winer, to be oublished. 8, 327 (1974). and references therein. Umpolung of Amine Reactivity. Nucleophilic a-(Secondary Amino)- New synthetic alky lation via Metalated Nitrosaminesr**lr”’l methods 0 By Dieter Seebach and Dieter Ended*] There are basically two kinds of hetero atoms in organic molecules: one kind confers electrophilic character upon the carbon atom to which it is bound, and the other kind turns it into a nucleophilic site. The development of methods permitting transitions between the two resulting categories of reagents has become an important task of modern organic synthesis. The scope of such umpolung of the reactivity of functional groups is discussed for the case of amines as an example. A method of preparing masked u-secondary amino carbanions consists in nitrosation of the secondary amine, followed by metalation of the resulting nitrosamine CL to the nitrogen, reaction with electrophiles, and subsequent denitrosation.