Neuronal Nitric Oxide Synthase Mediates Halothane- Induced Cerebral Microvascular Dilation Michael Staunton, M.B., F.F.A.R.C.S.I.,* Cathy Drexler, M.D.,† Phillip G
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Nitric Oxide in Health and Disease of the Nervous System H-Y Yun1,2, VL Dawson1,3,4 and TM Dawson1,3
Molecular Psychiatry (1997) 2, 300–310 1997 Stockton Press All rights reserved 1359–4184/97 $12.00 PROGRESS Nitric oxide in health and disease of the nervous system H-Y Yun1,2, VL Dawson1,3,4 and TM Dawson1,3 Departments of 1Neurology; 3Neuroscience; 4Physiology, Johns Hopkins University School of Medicine, Baltimore, MD, USA Nitric oxide (NO) is a widespread and multifunctional biological messenger molecule. It mediates vasodilation of blood vessels, host defence against infectious agents and tumors, and neurotransmission of the central and peripheral nervous systems. In the nervous system, NO is generated by three nitric oxide synthase (NOS) isoforms (neuronal, endothelial and immunologic NOS). Endothelial NOS and neuronal NOS are constitutively expressed and acti- vated by elevated intracellular calcium, whereas immunologic NOS is inducible with new RNA and protein synthesis upon immune stimulation. Neuronal NOS can be transcriptionally induced under conditions such as neuronal development and injury. NO may play a role not only in physiologic neuronal functions such as neurotransmitter release, neural development, regeneration, synaptic plasticity and regulation of gene expression but also in a variety of neurological disorders in which excessive production of NO leads to neural injury. Keywords: nitric oxide synthase; endothelium-derived relaxing factor; neurotransmission; neurotoxic- ity; neurological diseases Nitric oxide is probably the smallest and most versatile NO synthases isoforms and regulation of NO bioactive molecule identified. Convergence of multi- generation disciplinary efforts in the field of immunology, cardio- vascular pharmacology, chemistry, toxicology and neu- NO is formed by the enzymatic conversion of the guan- robiology led to the revolutionary novel concept of NO idino nitrogen of l-arginine by NO synthase (NOS). -
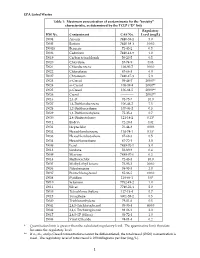
EPA Listed Wastes Table 1: Maximum Concentration of Contaminants For
EPA Listed Wastes Table 1: Maximum concentration of contaminants for the toxicity characteristic, as determined by the TCLP (D list) Regulatory HW No. Contaminant CAS No. Level (mg/L) D004 Arsenic 7440-38-2 5.0 D005 Barium 7440-39-3 100.0 D0018 Benzene 71-43-2 0.5 D006 Cadmium 7440-43-9 1.0 D019 Carbon tetrachloride 56-23-5 0.5 D020 Chlordane 57-74-9 0.03 D021 Chlorobenzene 108-90-7 100.0 D022 Chloroform 67-66-3 6.0 D007 Chromium 7440-47-3 5.0 D023 o-Cresol 95-48-7 200.0** D024 m-Cresol 108-39-4 200.0** D025 p-Cresol 106-44-5 200.0** D026 Cresol ------------ 200.0** D016 2,4-D 94-75-7 10.0 D027 1,4-Dichlorobenzene 106-46-7 7.5 D028 1,2-Dichloroethane 107-06-2 0.5 D029 1,1-Dichloroethylene 75-35-4 0.7 D030 2,4-Dinitrotoluene 121-14-2 0.13* D012 Endrin 72-20-8 0.02 D031 Heptachlor 76-44-8 0.008 D032 Hexachlorobenzene 118-74-1 0.13* D033 Hexachlorobutadiene 87-68-3 0.5 D034 Hexachloroethane 67-72-1 3.0 D008 Lead 7439-92-1 5.0 D013 Lindane 58-89-9 0.4 D009 Mercury 7439-97-6 0.2 D014 Methoxychlor 72-43-5 10.0 D035 Methyl ethyl ketone 78-93-3 200.0 D036 Nitrobenzene 98-95-3 2.0 D037 Pentachlorophenol 87-86-5 100.0 D038 Pyridine 110-86-1 5.0* D010 Selenium 7782-49-2 1.0 D011 Silver 7740-22-4 5.0 D039 Tetrachloroethylene 127-18-4 0.7 D015 Toxaphene 8001-35-2 0.5 D040 Trichloroethylene 79-01-6 0.5 D041 2,4,5-Trichlorophenol 95-95-4 400.0 D042 2,4,6-Trichlorophenol 88-06-2 2.0 D017 2,4,5-TP (Silvex) 93-72-1 1.0 D043 Vinyl Chloride 74-01-4 0.2 * Quantitation limit is greater than the calculated regulatory level. -

Differential Regulation of Endothelium Behavior by Progesterone and Medroxyprogesterone Acetate
P H CUTINI and others Progestins and vascular function 220:3 179–193 Research Differential regulation of endothelium behavior by progesterone and medroxyprogesterone acetate Pablo H Cutini1,2, Adria´n E Campelo1,2 and Virginia L Massheimer1,2 Correspondence should be addressed 1Ca´ tedra de Bioquı´mica Clı´nica II, Departamento de Biologı´a, Bioquı´mica y Farmacia, Universidad Nacional to V L Massheimer del Sur (UNS), San Juan 670, B8000ICN, Bahı´a Blanca, Argentina Email 2Consejo Nacional de Investigaciones Cientı´ficas y Te´ cnicas (CONICET), Argentina, Buenos Aires, Argentina [email protected] Abstract Medroxyprogesterone acetate (MPA) is a synthetic progestin commonly used in hormone Key Words replacement therapy (HRT). The aim of this research was to study and compare the effect of " cell migration progesterone (Pg) and MPA on the regulation of cellular events associated with vascular " medroxyprogesterone homeostasis and disease. Platelet adhesion to endothelial cells (ECs), nitric oxide (NO) acetate production, and cell migration were studied using murine ECs in vitro exposed to the " nitric oxide progestins. After 7 min of treatment, MPA significantly inhibited NO synthesis with respect " progesterone to control values; meanwhile, Pg markedly increased vasoactive production. In senile ECs, " vascular tissue the stimulatory action of Pg decreases; meanwhile, MPA maintained its ability to inhibit Journal of Endocrinology NO synthesis. The presence of RU486 antagonized the action of each steroid. When ECs were preincubated with PD98059 (MAPK inhibitor) or chelerythrine (protein kinase C (PKC) inhibitor) before Pg or MPA treatment, the former totally suppressed the steroid action, but the PKC antagonist did not affect NO production. -

Progesterone Using ALZET Osmotic Pumps
ALZET® Bibliography References on the Administration of Progesterone Using ALZET Osmotic Pumps Q8558: V. Joseph, et al. Progesterone decreases apnoea and reduces oxidative stress induced by chronic intermittent hypoxia in ovariectomized female rats. Exp Physiol 2020;105(6):1025-1034 Agents: Progesterone Vehicle: Cyclodextrin, 2-ß-Hydroxypropl-; Route: SC; Species: Rat; Pump: 2ML4; Duration: 28 days; ALZET Comments: Dose (4 mg/kg/day); Controls received mp w/ vehicle; animal info (Sprague-Dawley female rats (220-250g/57-70 days old)); post op. care (buprenorphine); Blood pressure measured via tail cuff method;93.3 mmHg - 105.2 mmHg;Progesterone aka prog; dependence; Q6232: S. F. Rosen, et al. T-Cell Mediation of Pregnancy Analgesia Affecting Chronic Pain in Mice. J Neurosci 2017;37(41):9819-9827 ALZET Comments: Estradiol, 17b-; Progesterone sulfate; SC; Mice; 2002; 14 days; Dose (17b-estradiol : 0.1 mg/kg/d, progesterone sulfate: 0.25 mg/kg/d, 0.1 mg/kg/d estradiol + 0.25 mg/kg/d progesterone); Controls received mp w/ vehicle; animal info (7-12 week old female C57BL/6J mice); replacement therapy (estradiol, ovariectomy); Therapeutic indication. Q6066: D. J. Morris, et al. Glucocorticoids and gut bacteria: "The GALF Hypothesis" in the metagenomic era. Steroids 2017;125(1-13 ALZET Comments: Chenodeoxycholic acid, progesterone, 11b-hydroxy-, corticosterone, deoxy-, corticosterone, 3α,5α-TH-, progesterone, 3α,5α-TH-11β-hydroxy-; SC; Rat; steroidal derivatives of corticosterone; Review presents the role of gut microbial metabolism of endogenous adrenocorticosteroids as a contributing factor in the etiology of essential hypertension. Q6204: S. McIlvride, et al. A progesterone-brown fat axis is involved in regulating fetal growth. -

Nitric Oxide Production, Inhibitory, Antioxidant and Antimycobacterial Activities of the Fruits Extract and Flavonoid Content of Schinus Terebinthifolius
Rev Bras Farmacogn 24(2014): 644-650 Original article Nitric oxide production, inhibitory, antioxidant and antimycobacterial activities of the fruits extract and flavonoid content of Schinus terebinthifolius Natalia R. Bernardesa, Marlon Heggdorne-Araújoa,b, Isabela F. J. C. Borgesb,c, Fabricio M. Almeidab, Eduardo P. Amaralb, Elena B. Lasunskaiab, Michelle F. Muzitanob,c,*, Daniela B. Oliveiraa,* aLaboratório de Tecnologia de Alimentos, Centro de Ciências e Tecnologias Agropecuárias, Universidade Estadual do Norte Fluminense Darcy Ribeiro, Campos dos Goytacazes, RJ, Brazil bLaboratório de Biologia do Reconhecer, Centro de Biociências e Biotecnologia, Universidade Estadual do Norte Fluminense Darcy Ribeiro, Campos dos Goytacazes, RJ, Brazil cLaboratório de Produtos Naturais, Curso de Farmácia, Universidade Federal do Rio de Janeiro, Campus Macaé, Polo Novo Cavaleiro, Instituto Macaé de Metrologia e Tecnologia, Macaé, RJ, Brazil ARTICLE INFO ABSTRACT Article history: The extract of the fruits from Schinus terebinthifolius Raddi, Anacardiaceae, was obtained by Received 28 May 2014 exhaustive extraction with methanol. Its fractions and isolated compounds were collected Accepted 16 October 2014 by fractionation with RP-2 column chromatography. The crude extract, the flavonoid frac- tion and the isolated compound identified as apigenin (1), were investigated regarding its Keywords: inhibitory action of nitric oxide production by LPS-stimulated macrophages, antioxidant Anacardiaceae activity by DPPH and the antimycobacterial activity against Mycobacterium bovis BCG. The Apigenin samples exhibited a significant inhibitory effect on the nitric oxide production (e.g., 1, IC50 Inflammation 19.23 ± 1.64 μg/ml) and also showed antioxidant activity. In addition, S. terebinthifolius sam- Mycobacterium ples inhibited the mycobacterial growth (e.g., 1, IC50 14.53 ± 1.25 μg/ml). -

A STUDY of the COMPOSITION and SOME REACTIONS OP the ETHERATE OP ALUMINUM TRIBTHYL DISSERTATION. Presented in Partial Ïtilfillm
A STUDY OF THE COMPOSITION AND SOME REACTIONS OP THE ETHERATE OP ALUMINUM TRIBTHYL DISSERTATION. Presented in Partial ïtilfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State ■ University ■ By EVELYN RUTH BLUST BAKER, B.A., 1.5, w The Ohio State University 1953 Approved by* Z’ f ' X ' / - y 1 ■■ ' . 7 ' ■------- 4 .dviser ACKNOWLEDGEMENT The author would like to thank Dr. Harry fl. Sisler for suggesting this study and for his continued interest and guidance throughout the course of the work. 933320 ii TABLE OF CONTENTS Page Chapter I* Introduction A» Purpose and Scope of this Research 1 B. Review of Methods of Preparation of Aluminum Triethyl and Aluminum Triethyl Ethorate 2 C. Properties Given in the Literature 4 Chapter II. Experimental Results and Discussion A. Experimental Methods and Precautions Employed in this Work 5 B. Preparation of Aluminum Triethyl and Aluminum Triethyl Etherate 8 0 . A Phase Study of the Binary System Aluminum Triethyl-Diethyl Ether 11 Do Reaction of Aluminum Triethyl Etherate with Alkali Metal Alkyls 24 E. Reaction of Aluminum Triethyl Etherate with Sulfur Dioxide 30 P. Reaction of Aluminum Triethyl Etherate with Nitrogen Dioxide 35 6 . Reaction of Aluminum Triethyl Etherate with Nitric Oxide 42 H. Reaction of Aluminum Triethyl Etherate with Pyridine 47 Ill I. Additional Reactions of Aluminum Triethyl Etherate* 1 . Reactionwith Carbon Dioxide 50 2. Reactionwith Oxygen 51 3 . Reaction with Sulfur Trioxide 53 4 * Reaction with Carbon Tetrachloride 54 Chapter III. Conclusions and Summary ^6 Bibliography 59 A STUDY OP THE COMPOSITION AND SOME REACTIONS OP THE ETHERATE OP ALUMINUM TRIETHYL CHAPTER I. -

EUROPEAN PHARMACOPOEIA 10.0 Index 1. General Notices
EUROPEAN PHARMACOPOEIA 10.0 Index 1. General notices......................................................................... 3 2.2.66. Detection and measurement of radioactivity........... 119 2.1. Apparatus ............................................................................. 15 2.2.7. Optical rotation................................................................ 26 2.1.1. Droppers ........................................................................... 15 2.2.8. Viscosity ............................................................................ 27 2.1.2. Comparative table of porosity of sintered-glass filters.. 15 2.2.9. Capillary viscometer method ......................................... 27 2.1.3. Ultraviolet ray lamps for analytical purposes............... 15 2.3. Identification...................................................................... 129 2.1.4. Sieves ................................................................................. 16 2.3.1. Identification reactions of ions and functional 2.1.5. Tubes for comparative tests ............................................ 17 groups ...................................................................................... 129 2.1.6. Gas detector tubes............................................................ 17 2.3.2. Identification of fatty oils by thin-layer 2.2. Physical and physico-chemical methods.......................... 21 chromatography...................................................................... 132 2.2.1. Clarity and degree of opalescence of -

Top 100 Chemicals List
CAS No. Chemical Name Trade or Common Name Synonyms & Notes Fire Code Haz Class 71556 1,1,1-TRICHLOROETHANE TRICHLOR 111 DEGRS COLD/V BRAKLEEN #5090 IRR,OHH 71556 1,1,1-TRICHLOROETHANE TRI - ETHANE (R) 377 LO VOC SATIN LACQUER IRR,OHH 71556 1,1,1-TRICHLOROETHANE SAFETY KLEEN 105 SOLVENT METHYL CHLOROFORM IRR,OHH 71556 1,1,1-TRICHLOROETHANE YELLOW LO VOC -TRAFFIC PAINT LACQUER THINNER IRR,OHH 127184 1,1,2,2,TETRACHLOROETHYLENE WASTE PERCHLOROETHYLENE BRAKE CLEAN CAR,IRR,OHH,SENS 127184 1,1,2,2,TETRACHLOROETHYLENE PERC CHLORINATED HYDROCARBON CAR,IRR,OHH,SENS 107062 1,2-DICHLOROETHANE ETHYLENE DICHLORIDE CAR,FLIB,IRR,OHH 57556 1,2-PROPANEDIOL RUBIGAN A.S. FUNGICIDE MURPHY OIL SOAP CLIIIB,IRR,OHH 57556 1,2-PROPANEDIOL HD HEAT TRANSFER FLUID PROPYLENE GLYCOL CLIIIB,IRR,OHH 57556 1,2-PROPANEDIOL DOW FROST GLYCOL LATEX COATINGS- WATERBORNE CLIIIB,IRR,OHH 78933 2-BUTANONE METHYL ETHYL KETONE WASTE PAINT FILTERS & DEBRIS FLIB,IRR,OHH 78933 2-BUTANONE LACQUER THINNER MEK FLIB,IRR,OHH 78933 2-BUTANONE BURKE 3203 PART A PRIMER PART B FLIB,IRR,OHH 64197 ACETIC ACID VINEGAR ACETIC ACID, <10% IRR,OHH 64197 ACETIC ACID FIXER, REPLENISHER, DEVELOPER ACETIC ACID, >12% COR,OHH 64197 ACETIC ACID FIXER, REPLENISHER, DEVELOPER ACETIC ACID, 99-100% CLII,COR,OHH 67641 ACETONE THINNER WASTE ACETONE/HEXANE FLIB,IRR,OHH 67641 ACETONE BRAKE CLEANER 2-PROPANONE, LIQUID FLIB,IRR,OHH 67641 ACETONE DIMETHYL KETONE FLIB,IRR,OHH 74862 ACETYLENE ACETYLEN, GAS WELDING GAS FLG,UR1 74862 ACETYLENE ACETYLENE - ETHYNE FLG,UR1 79061 ACRYLAMIDE ACRYLAMIDE, CRYSTALS P - 4 CAR,IRR,OHH,TOX,UR2 7664417 AMMONIA ANHYDROUS AMMONIA ANHYDROUS AMMONIA COR 7664417 AMMONIA LESS THAN 1% AQUA AMMONIA NON HAZ 7664417 AMMONIA 50% AQUA AMMONIA COR,TOX 7664417 AMMONIA GREATER THAN 30% AQUA AMMONIA TOX, IRR CAS No. -

Nitrogen Oxides (Nox), Why and How They Are Controlled
United States Office of Air Quality EPA 456/F-99-006R Environmental Protection Planning and Standards November 1999 Agency Research Triangle Park, NC 27711 Air EPA-456/F-99-006R November 1999 Nitrogen Oxides (NOx), Why and How They Are Controlled Prepared by Clean Air Technology Center (MD-12) Information Transfer and Program Integration Division Office of Air Quality Planning and Standards U.S. Environmental Protection Agency Research Triangle Park, North Carolina 27711 DISCLAIMER This report has been reviewed by the Information Transfer and Program Integration Division of the Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency and approved for publication. Approval does not signify that the contents of this report reflect the views and policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products is not intended to constitute endorsement or recommendation for use. Copies of this report are available form the National Technical Information Service, U.S. Department of Commerce, 5285 Port Royal Road, Springfield, Virginia 22161, telephone number (800) 553-6847. CORRECTION NOTICE This document, EPA-456/F-99-006a, corrects errors found in the original document, EPA-456/F-99-006. These corrections are: Page 8, fourth paragraph: “Destruction or Recovery Efficiency” has been changed to “Destruction or Removal Efficiency;” Page 10, Method 2. Reducing Residence Time: This section has been rewritten to correct for an ambiguity in the original text. Page 20, Table 4. Added Selective Non-Catalytic Reduction (SNCR) to the table and added acronyms for other technologies. Page 29, last paragraph: This paragraph has been rewritten to correct an error in stating the configuration of a typical cogeneration facility. -

The Neuronal Nitric Oxide Synthase Gene Is Critically Involved in Neurobehavioral Effects of Alcohol
The Journal of Neuroscience, October 1, 2002, 22(19):8676–8683 The Neuronal Nitric Oxide Synthase Gene Is Critically Involved in Neurobehavioral Effects of Alcohol Rainer Spanagel,1,2 So¨ ren Siegmund,1,3 Michael Cowen,1 Karl-Christian Schroff,1 Gunter Schumann,1 Magdalena Fiserova,4 Inge Sillaber,2 Stefan Wellek,5 Manfred Singer,3 and Jo¨ rg Putzke6 1Department of Psychopharmacology, Central Institute of Mental Health (CIMH), University of Heidelberg, 68159 Mannheim, Germany, 2Drug Abuse Research Group, Max Planck Institute of Psychiatry, 80804 Munich, Germany, 3Department of Medicine IV, University Hospital of Heidelberg, 68167 Mannheim, Germany, 4Institute of Pharmacology, Charles University, 10000 Prague, Czech Republic, 5Department of Biostatistics, CIMH, 68159 Mannheim, Germany, and 6Department of Medical Neurobiology, University of Magdeburg, 39120 Magdeburg, Germany In the present study, we describe a new role of the neuronal thermia, although plasma ethanol concentrations were not sig- nitric oxide synthase (nNOS) gene in the regulation of alcohol nificantly different from those of controls. Our findings contrast drinking behavior. Mice deficient in the nNOS gene (nNOS Ϫ/Ϫ) with previous reports that showed that nonselective NOS in- and wild-type control mice were submitted to a two-bottle hibitors decrease alcohol consumption. However, because al- free-choice procedure with either water or increasing concen- cohol consumption was suppressed in wild-type as well as G trations of alcohol (2–16%) for 6 weeks. nNOS Ϫ/Ϫ mice did nNOS Ϫ/Ϫ mice by the NOS inhibitor N -nitro-L-arginine not differ in consumption and preference for low alcohol con- methyl ester, we conclude that the effect of nonselective NOS centrations from wild-type animals; however, nNOS Ϫ/Ϫ mice inhibitors on alcohol drinking is not mediated by nNOS. -

Nitric Oxide Synthase Acutely Regulates Progesterone Production by in Vitro Cultured Rabbit Corpora Lutea
275 Nitric oxide synthase acutely regulates progesterone production by in vitro cultured rabbit corpora lutea A Gobbetti, C Boiti1, C Canali1 and M Zerani Dipartimento di Biologia Molecolare, Cellulare e Animale, Universita` di Camerino, Camerino, Italy 1Istituto di Fisiologia Veterinaria, Universita` di Perugia, Perugia, Italy (Requests for offprints should be addressed to C Boiti, Istituto di Fisiologia Veterinaria, Universita` di Perugia, via S Costanzo 4, 06100 Perugia, Italy) Abstract We examined the presence and the regulation of nitric -NAME. All treatments failed to modify basal androgens oxide (NO) synthase (NOS) using in vitro cultured corpora and 17â-oestradiol was not detectable in either control or lutea (CL) obtained from rabbits at days 4 and 9 of treated CL. These results suggest that NO is effectively pseudopregnancy. The role of NO and NOS on steroido- involved in the regulation process of steroidogenesis, genesis was also investigated using the same CL prepara- independently of 17â-oestradiol. PGF-2á had no effect on tions after short-term incubations (30 min and 2 h) with day 4, but induced luteolysis on day 9, by reducing the NO donor, sodium nitroprusside (NP), the NOS progesterone (Pc0·01) to about 18% of control. The inhibitor, Nù-nitro--arginine methyl ester (-NAME) luteolytic action of PGF-2á was completely reversed and prostaglandin (PG) F-2á. The basal NOS activity was by co-incubation with -NAME, thus supporting the greater in CL at day 4 than at day 9, and was also hypothesis that luteolysis is mediated by NO. The addition differently modulated by PGF-2á, depending on the age of NP or -NAME did not modify the in vitro release of of the CL. -

(2020) . Can Inhaling Nitric Oxide Treat — Or Prevent — COVID-19? MGH Wants to Find Out
Costa, J. (2020) . Can inhaling nitric oxide treat — Or prevent — COVID-19? MGH Wants To Find Out. Massachusetts General Hospital. (Jesse Costa/WBUR) Abstract: Researchers at Massachusetts General Hospital (MGH) are studying whether inhaling a gas called nitric oxide can help treat patients with COVID-19, or even prevent people from getting the disease. Although nitric oxide is widely used to treat patients with respiratory failure, MGH is the only hospital in the United States, and one of the few in the world, studying it for the treatment of COVID-19, according to the CDC. (MGH is conducting one of the studies in cooperation with teams in Alabama and Louisiana. Nitric oxide is a colorless gas, not to be confused with "nitrous oxide," better known as laughing gas, or "nitrogen dioxide," a smoggy brown air pollutant. The human body makes nitric oxide on its own, and uses it as a chemical messenger. Low levels of nitric oxide constrict blood vessels; higher levels dilate them, increasing blood flow. When patients inhale it through a face mask or mechanical ventilator, it can open airways and help them breathe better. Libby Hohmann, a physician in the infectious diseases division at Massachusetts General Hospital, calls nitric oxide "a favorite of MGH.It has advantageous effects for people with certain kinds of respiratory failure," says Hohmann, "and it also appears to have some direct antiviral effects."The inhaled gas only affects the lungs, and the effects are short-lived. "You see an effect within minutes,"says Robert Kacmarek, director of respiratory care at MGH and one of the leaders of the nitric oxide trials there.