A New Computational Approach to Evaluating Systemic Gene–Gene Interactions in a Pathway Affected by Drug LY294002
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Brief Genetics Report Haplotype Structures and Large
Brief Genetics Report Haplotype Structures and Large-Scale Association Testing of the 5 AMP-Activated Protein Kinase Genes PRKAA2, PRKAB1, and PRKAB2 With Type 2 Diabetes Maria W. Sun,1,2 Jennifer Y. Lee,1,2 Paul I.W. de Bakker,1,2,3 Noe¨l P. Burtt,2 Peter Almgren,4 Lennart Råstam,5 Tiinamaija Tuomi,6 Daniel Gaudet,7 Mark J. Daly,2,8 Joel N. Hirschhorn,2,3,9 David Altshuler,1,2,3,8,10 Leif Groop,4,6 and Jose C. Florez1,2,8,10 AMP-activated protein kinase (AMPK) is a key molecular plasma glucose, or insulin sensitivity. Several nominal asso- regulator of cellular metabolism, and its activity is induced ciations of variants in PRKAA2 and PRKAB1 with BMI appear by both metformin and thiazolidinedione antidiabetic med- to be consistent with statistical noise. Diabetes 55:849–855, ications. It has therefore been proposed both as a putative 2006 agent in the pathophysiology of type 2 diabetes and as a valid target for therapeutic intervention. Thus, the genes that encode the various AMPK subunits are intriguing ype 2 diabetes arises from the complex interplay candidates for the inherited basis of type 2 diabetes. We therefore set out to test for the association of common of various pathophysiologic mechanisms involv- variants in the genes that encode three selected AMPK ing peripheral insulin resistance and relative subunits with type 2 diabetes and related phenotypes. Of Tinsulin insufficiency. The final expression of the the seven genes that encode AMPK isoforms, we initially diabetic phenotype is strongly influenced by inheritance; chose PRKAA2, PRKAB1, and PRKAB2 because of their however, with the exception of rare monogenic forms of higher prior probability of association with type 2 diabetes, diabetes, common type 2 diabetes is thought to have a based on previous reports of genetic linkage, functional polygenic architecture (1). -

Association Study of AMP-Activated Protein Kinase Subunit Genes In
European Journal of Endocrinology (2009) 161 405–409 ISSN 0804-4643 CLINICAL STUDY Association study of AMP-activated protein kinase subunit genes in polycystic ovary syndrome Kari Sproul1,2, Michelle R Jones3, Ricardo Azziz1,2,4 and Mark O Goodarzi1,3,4,5 1Department of Obstetrics and Gynecology, Cedars-Sinai Medical Center, Los Angeles, California 90048, USA, 2Department of Obstetrics and Gynecology, the David Geffen School of Medicine at UCLA, Los Angeles, California 90095, USA, 3Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, Cedars-Sinai Medical Center, 8700 Beverly Boulevard, Room B-131, Los Angeles, California 90048, USA, 4Department of Medicine, the David Geffen School of Medicine at UCLA, Los Angeles, California 90095, USA and 5Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, California 90048, USA (Correspondence should be addressed to M O Goodarzi at Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, Cedars-Sinai Medical Center; Email: [email protected]) Abstract Objective: To examine the genes for AMP-activated protein kinase (AMPK) subunits a2(PRKAA2) and g3(PRKAG3) as candidates for polycystic ovary syndrome (PCOS) and its component traits. Design and methods: A total of 287 white PCOS women were recruited from the reproductive endocrinology clinic at the University of Alabama at Birmingham and 187 white control subjects were recruited from the surrounding community. Seven PRKAA2 single nucleotide polymorphisms (SNPs) and four PRKAG3 SNPs were genotyped in PCOS cases and controls. Genotyping and association analysis were performed at Cedars-Sinai Medical Center. Results: Nominal associations of PRKAA2 variants with insulin-related traits and the PRKAG3 Pro71Ala variant with PCOS were not statistically significant after multiple testing correction. -
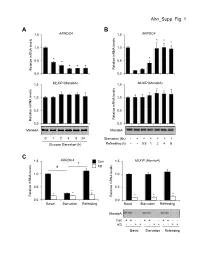
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

The Study of Copy Number Variations in the Regions of PRKAB2 and PPM1K Among Congenital Heart Defects Patients
The study of copy number variations in the regions of PRKAB2 and PPM1K among congenital heart defects patients Han-Quan Dong1 Yue-Xin Du2 1. Department of Pneumology, Tianjin Children’s Hospital, Tianjin, 300074–China 2. Department of Child Healthcare, Tianjin Municipal Women and Children health care center, Tianjin, 300070, China http://dx.doi.org/10.1590/1806-9282.65.6.786 SUMMARY OBJECTIVE: This study was to assess the genetic association of copy number variations in two genes (PRKAB2 and PPM1K) located in two regions (tetralogy of Fallot and ventricular septal defect) in a Chinese Han population. METHODS: A total of 200 congenital heart disease patients (100 tetralogy of Fallot patients and 100 ventricular septal defect patients) and 100 congenital heart defect-free controls were recruited, and quantitative real-time PCR analysis was used to replicate the asso- ciation of two copy number variations with congenital heart defects in a Chinese Han population. RESULTS: One deletion at PRKAB2 and one duplication at PPM1K were found in two of the tetralogy of Fallot patients, respectively; while all these regions were duplicated in both ventricular septal defect patients and in the 100 congenital heart defects-free controls. CONCLUSIONS: We replicated the copy number variations at the disease-candidate genes of PRKAB2 and PPM1K with tetralogy of Fallot in a Chinese Han population, and in patients with ventricular septal defect mutations in these two genes were not found. These results indicate the same molecular population genetics exist in these two genes with different ethnicity. This shows that these two genes are possibly specific pf tetralogy of Fallot candidates. -
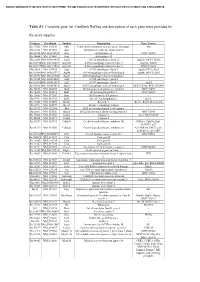
Table S1. Complete Gene List. Genbank Refseq and Description of Each Gene Were Provided By
Document downloaded from http://www.elsevier.es, day 24/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Table S1. Complete gene list. GenBank RefSeq and description of each gene were provided by the array supplier. Unigene GeneBank Symbol Description Gene Name/s Rn.11422 NM_033230 Akt1 V-akt murine thymoma viral oncogene homolog 1 Akt Rn.2104 NM_019288 App Amyloid beta (A4) precursor protein - Rn.23323 NM_001034933 Arsa Arylsulfatase A MGC125207 Rn.94004 NM_033443 Arsb Arylsulfatase B - Rn.6224 NM_001038495 Atg12 ATG12 autophagy related 12 Apg12l, MGC125080 Rn.101734NM_001108809 Atg16l1 ATG16 autophagy related 16-like 1 Apg16l, Wdr30 Rn.104199NM_001191560 Atg16l2 ATG16 autophagy related 16-like 2 RGD1311400 Rn.3084 NM_134394 Atg3 ATG3 autophagy related 3 Apg3l, PIG-1, Pig1 Rn.163086NM_001025711 Atg4b ATG4 autophagy related 4 homolog B Apg4b, MGC112887 Rn.23378 NM_001107948 Atg4c ATG4 autophagy related 4 homolog C - Rn.98385 NM_001014250 Atg5 ATG5 autophagy related 5 - Rn.162765NM_001012097 Atg7 ATG7 autophagy related 7 Apg7l Rn.35248 NM_001014218 Atg9a ATG9 autophagy related 9 homolog A MGC105908, RGD1310450 Rn.36696 NM_022698 Bad BCL2-associated agonist of cell death MGC72439 Rn.14598 NM_053812 Bak1 BCL2-antagonist/killer 1 MGC108627 Rn.10668 NM_017059 Bax Bcl2-associated X protein - Rn.9996 NM_016993 Bcl2 B-cell CLL/lymphoma 2 Bcl-2 Rn.10323 NM_031535 Bcl2l1 Bcl2-like 1 Bcl-xl, Bcl2l, Bclx, bcl-X Rn.2776 NM_053739 Becn1 Beclin 1, autophagy related - Rn.31142 NM_022684 -

(AMP)-Activated Protein Kinase: a New Target for Nutraceutical Compounds
International Journal of Molecular Sciences Review Adenosine Monophosphate (AMP)-Activated Protein Kinase: A New Target for Nutraceutical Compounds Fabiola Marín-Aguilar 1, Luis E. Pavillard 1, Francesca Giampieri 2, Pedro Bullón 1 and Mario D. Cordero 1,* 1 Research Laboratory, Oral Medicine Department, University of Sevilla, Sevilla 41009, Spain; [email protected] (F.M.-A.); [email protected] (L.E.P.); [email protected] (P.B.) 2 Dipartimento di Scienze Cliniche Specialistiche ed Odontostomatologiche—Sez. Biochimica, Università Politecnica delle Marche, Ancona 60100, Italy; [email protected] * Correspondence: [email protected]; Tel.: +34-954-481-120; Fax: +34-954-486-784 Academic Editor: Rosa M. Lamuela-Raventós Received: 17 November 2016; Accepted: 23 January 2017; Published: 29 January 2017 Abstract: Adenosine monophosphate-activated protein kinase (AMPK) is an important energy sensor which is activated by increases in adenosine monophosphate (AMP)/adenosine triphosphate (ATP) ratio and/or adenosine diphosphate (ADP)/ATP ratio, and increases different metabolic pathways such as fatty acid oxidation, glucose transport and mitochondrial biogenesis. In this sense, AMPK maintains cellular energy homeostasis by induction of catabolism and inhibition of ATP-consuming biosynthetic pathways to preserve ATP levels. Several studies indicate a reduction of AMPK sensitivity to cellular stress during aging and this could impair the downstream signaling and the maintenance of the cellular energy balance and the stress resistance. However, several diseases have been related with an AMPK dysfunction. Alterations in AMPK signaling decrease mitochondrial biogenesis, increase cellular stress and induce inflammation, which are typical events of the aging process and have been associated to several pathological processes. -

PRKAA2 (Human) Recombinant Protein (Q01)
PRKAA2 (Human) Recombinant that monitors cellular energy status. In response to Protein (Q01) cellular metabolic stresses, AMPK is activated, and thus phosphorylates and inactivates acetyl-CoA carboxylase Catalog Number: H00005563-Q01 (ACC) and beta-hydroxy beta-methylglutaryl-CoA reductase (HMGCR), key enzymes involved in regulating Regulation Status: For research use only (RUO) de novo biosynthesis of fatty acid and cholesterol. Studies of the mouse counterpart suggest that this Product Description: Human PRKAA2 partial ORF ( catalytic subunit may control whole-body insulin NP_006243, 453 a.a. - 552 a.a.) recombinant protein sensitivity and is necessary for maintaining myocardial with GST-tag at N-terminal. energy homeostasis during ischemia. [provided by RefSeq] Sequence: MSLQLYLVDNRSYLLDFKSIDDEVVEQRSGSSTPQRS CSAAGLHRPRSSFDSTTAESHSLSGSLTGSLTGSTLS SVSPRLGSHTMDFFEMCASLITTLAR Host: Wheat Germ (in vitro) Theoretical MW (kDa): 36.74 Applications: AP, Array, ELISA, WB-Re (See our web site product page for detailed applications information) Protocols: See our web site at http://www.abnova.com/support/protocols.asp or product page for detailed protocols Preparation Method: in vitro wheat germ expression system Purification: Glutathione Sepharose 4 Fast Flow Storage Buffer: 50 mM Tris-HCI, 10 mM reduced Glutathione, pH=8.0 in the elution buffer. Storage Instruction: Store at -80°C. Aliquot to avoid repeated freezing and thawing. Entrez GeneID: 5563 Gene Symbol: PRKAA2 Gene Alias: AMPK, AMPK2, PRKAA Gene Summary: The protein encoded by this gene is a catalytic subunit of the AMP-activated protein kinase (AMPK). AMPK is a heterotrimer consisting of an alpha catalytic subunit, and non-catalytic beta and gamma subunits. AMPK is an important energy-sensing enzyme Page 1/1 Powered by TCPDF (www.tcpdf.org). -

AMPK Causes Cell Cycle Arrest in LKB1-Deficient Cells Via Activation of CAMKK2
Published OnlineFirst May 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0479 Cell Cycle and Senescence Molecular Cancer Research AMPK Causes Cell Cycle Arrest in LKB1-Deficient Cells via Activation of CAMKK2 Sarah Fogarty, Fiona A. Ross, Diana Vara Ciruelos, Alexander Gray, Graeme J. Gowans, and D. Grahame Hardie Abstract The AMP-activated protein kinase (AMPK) is activated by also caused G1 arrest similar to that caused by expression of LKB1, phosphorylation at Thr172, either by the tumor suppressor kinase while expression of a dominant-negative AMPK mutant, or a þ LKB1 or by an alternate pathway involving the Ca2 /calmodulin- double knockout of both AMPK-a subunits, also prevented the dependent kinase, CAMKK2. Increases in AMP:ATP and ADP:ATP cell-cycle arrest caused by A23187. These mechanistic findings ratios, signifying energy deficit, promote allosteric activation and confirm that AMPK activation triggers cell-cycle arrest, and also net Thr172 phosphorylation mediated by LKB1, so that the LKB1– suggest that the rapid proliferation of LKB1-null tumor cells is due AMPK pathway acts as an energy sensor. Many tumor cells carry to lack of the restraining influence of AMPK. However, cell-cycle loss-of-function mutations in the STK11 gene encoding LKB1, but arrest can be restored by reexpressing LKB1 or a constitutively LKB1 reexpression in these cells causes cell-cycle arrest. Therefore, active CAMKK2, or by pharmacologic agents that increase intra- þ it was investigated as to whether arrest by LKB1 is caused by cellular Ca2 and thus activate endogenous CAMKK2. activation of AMPK or of one of the AMPK-related kinases, which are also dependent on LKB1 but are not activated by CAMKK2. -

Association of AMP-Activated Protein Kinase with Risk and Progression of Non–Hodgkin Lymphoma
Published OnlineFirst February 8, 2013; DOI: 10.1158/1055-9965.EPI-12-1014 Cancer Epidemiology, Research Article Biomarkers & Prevention Association of AMP-Activated Protein Kinase with Risk and Progression of Non–Hodgkin Lymphoma Aaron E. Hoffman1, Kathryn Demanelis1, Alan Fu2, Tongzhang Zheng2, and Yong Zhu2 Abstract Background: Metabolic dysregulation has been identified as an "emerging hallmark" of cancer. The heterotrimeric AMP-activated protein kinase (AMPK) complex is a central regulator of the metabolic system and an important component of the mTOR pathway and the p53 axis, making it uniquely positioned to influence carcinogenesis through its canonical functions in the metabolic arena, as well as through more traditional mechanisms such as regulation of apoptosis and angiogenesis. Methods: We conducted a population-based genetic association study to examine the impact of mutations in AMPK subunit genes on risk of non–Hodgkin lymphoma (NHL). We also analyzed public microarray data to determine the expression of AMPK in NHL cells and to assess the influence of AMPK expression on overall survival in patients with NHL. Results: We identified an AMPK subunit haplotype, which was significantly associated with NHL [OR, 5.44, 95% confidence interval (CI), 2.15–13.75] in women with no family history of cancer. Haplotypes in two subunits, PRKAA2 and PRKAG3, were nominally associated with the follicular and diffuse large B-cell lymphoma histologic subtypes, respectively, although these associations did not retain statistical significance after correction for multiple comparisons. Further, both of these subunits were differentially expressed (P < 0.05) in one or more lymphoma cell type, and higher expression of two versions of the AMPK-b subunit was significantly associated with increased 5-year survival among patients with NHL (P ¼ 0.001 and P ¼ 0.021). -

Therapeutic Inhibition of Mir-802 Protects Against Obesity Through
Supporting Information Therapeutic inhibition of miR-802 protects against obesity through AMPK-mediated regulation of hepatic lipid metabolism Yangyue Ni †, Zhipeng Xu †, Chen Li †, Yuxiao Zhu, Ran Liu, Fan Zhang, Hao Chang, Maining Li, Liang Sheng, Zhong Li, Min Hou, Lin Chen, Hong You, Donald P. McManus, Wei Hu, Yinong Duan, Yu Liu, Minjun Ji * † Yangyue Ni, Zhipeng Xu and Chen Li contributed equally to this work. 1 Figure S1. Schistosoma japonicum infection improves lipid metabolism in HFD mice. (A) Sixteen 10-week-old male C57BL/6 mice were separated equally into two groups and fed a normal diet. Each mouse in the ND-inf group was infected with 10 ± 1 S. 2 japonicum cercariae and the ND-con group was established as the normal control group. Sixteen 6-week-old male C57BL/6 mice that had been maintained on a high-fat diet for 1 month were randomly divided into two groups: a high-fat diet-chronic infection group (HFD-inf) and the control group (HFD-con). Mice were sacrificed 9 weeks after the infection. (B) Dynamic changes in body weight of ND- and HFD-fed mice after infection with Schistosoma japonicum. (C) Cholesterol, TG, HDL-C, LDL-C levels in sera of HFD mice upon infection. (D, E) Representative images of liver sections from ND-con, HFD-con, ND-inf or HFD-inf mice stained with Oil red O. (F) qRT-PCR quantification of lipid-related genes expression in livers of four groups of mice (ND- con, HFD-con, ND-inf or HFD-inf). (G) qRT-PCR quantification of lipid-related gene expression in adipose tissues of three groups of mice (ND-con, HFD-con and HFD-inf). -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain