AMPK Causes Cell Cycle Arrest in LKB1-Deficient Cells Via Activation of CAMKK2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![FK506-Binding Protein 12.6/1B, a Negative Regulator of [Ca2+], Rescues Memory and Restores Genomic Regulation in the Hippocampus of Aging Rats](https://docslib.b-cdn.net/cover/6136/fk506-binding-protein-12-6-1b-a-negative-regulator-of-ca2-rescues-memory-and-restores-genomic-regulation-in-the-hippocampus-of-aging-rats-16136.webp)
FK506-Binding Protein 12.6/1B, a Negative Regulator of [Ca2+], Rescues Memory and Restores Genomic Regulation in the Hippocampus of Aging Rats
This Accepted Manuscript has not been copyedited and formatted. The final version may differ from this version. A link to any extended data will be provided when the final version is posted online. Research Articles: Neurobiology of Disease FK506-Binding Protein 12.6/1b, a negative regulator of [Ca2+], rescues memory and restores genomic regulation in the hippocampus of aging rats John C. Gant1, Eric M. Blalock1, Kuey-Chu Chen1, Inga Kadish2, Olivier Thibault1, Nada M. Porter1 and Philip W. Landfield1 1Department of Pharmacology & Nutritional Sciences, University of Kentucky, Lexington, KY 40536 2Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294 DOI: 10.1523/JNEUROSCI.2234-17.2017 Received: 7 August 2017 Revised: 10 October 2017 Accepted: 24 November 2017 Published: 18 December 2017 Author contributions: J.C.G. and P.W.L. designed research; J.C.G., E.M.B., K.-c.C., and I.K. performed research; J.C.G., E.M.B., K.-c.C., I.K., and P.W.L. analyzed data; J.C.G., E.M.B., O.T., N.M.P., and P.W.L. wrote the paper. Conflict of Interest: The authors declare no competing financial interests. NIH grants AG004542, AG033649, AG052050, AG037868 and McAlpine Foundation for Neuroscience Research Corresponding author: Philip W. Landfield, [email protected], Department of Pharmacology & Nutritional Sciences, University of Kentucky, 800 Rose Street, UKMC MS 307, Lexington, KY 40536 Cite as: J. Neurosci ; 10.1523/JNEUROSCI.2234-17.2017 Alerts: Sign up at www.jneurosci.org/cgi/alerts to receive customized email alerts when the fully formatted version of this article is published. -

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Brief Genetics Report Haplotype Structures and Large
Brief Genetics Report Haplotype Structures and Large-Scale Association Testing of the 5 AMP-Activated Protein Kinase Genes PRKAA2, PRKAB1, and PRKAB2 With Type 2 Diabetes Maria W. Sun,1,2 Jennifer Y. Lee,1,2 Paul I.W. de Bakker,1,2,3 Noe¨l P. Burtt,2 Peter Almgren,4 Lennart Råstam,5 Tiinamaija Tuomi,6 Daniel Gaudet,7 Mark J. Daly,2,8 Joel N. Hirschhorn,2,3,9 David Altshuler,1,2,3,8,10 Leif Groop,4,6 and Jose C. Florez1,2,8,10 AMP-activated protein kinase (AMPK) is a key molecular plasma glucose, or insulin sensitivity. Several nominal asso- regulator of cellular metabolism, and its activity is induced ciations of variants in PRKAA2 and PRKAB1 with BMI appear by both metformin and thiazolidinedione antidiabetic med- to be consistent with statistical noise. Diabetes 55:849–855, ications. It has therefore been proposed both as a putative 2006 agent in the pathophysiology of type 2 diabetes and as a valid target for therapeutic intervention. Thus, the genes that encode the various AMPK subunits are intriguing ype 2 diabetes arises from the complex interplay candidates for the inherited basis of type 2 diabetes. We therefore set out to test for the association of common of various pathophysiologic mechanisms involv- variants in the genes that encode three selected AMPK ing peripheral insulin resistance and relative subunits with type 2 diabetes and related phenotypes. Of Tinsulin insufficiency. The final expression of the the seven genes that encode AMPK isoforms, we initially diabetic phenotype is strongly influenced by inheritance; chose PRKAA2, PRKAB1, and PRKAB2 because of their however, with the exception of rare monogenic forms of higher prior probability of association with type 2 diabetes, diabetes, common type 2 diabetes is thought to have a based on previous reports of genetic linkage, functional polygenic architecture (1). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

32-3099: PRKAB1 Recombinant Protein Description
9853 Pacific Heights Blvd. Suite D. San Diego, CA 92121, USA Tel: 858-263-4982 Email: [email protected] 32-3099: PRKAB1 Recombinant Protein Alternative Name : AMPK,HAMPKb,5'-AMP-activated protein kinase subunit beta-1,AMPK subunit beta-1,AMPKb,PRKAB1. Description Source : E.coli. PRKAB1 Human Recombinant produced in E.Coli is a single, non-glycosylated, polypeptide chain containing 293 amino acids (1-270 a.a.) and having a molecular mass of 32.8 kDa. The PRKAB1 is fused to a 23 amino acid His Tag at N- Terminus and purified by proprietary chromatographic techniques. 5'-AMP-activated protein kinase subunit beta-1 (PRKAB1) hinders protein, carbohydrate and lipid biosynthesis, in addition to cell growth and proliferation. AMPK is a heterotrimer comprised of an alpha catalytic subunit, and non-catalytic beta and gamma subunits. AMPK acts via direct phosphorylation of metabolic enzymes, and longer-term effects by phosphorylation of transcription regulators. PRKAB1 is a regulator of cellular polarity by remodeling the actin cytoskeleton; most likely by indirectly activating myosin. Beta non-catalytic subunit acts as a scaffold on which the AMPK complex compiles, through its C-terminus that joins alpha (PRKAA1 or PRKAA2) and gamma subunits (PRKAG1, PRKAG2 or PRKAG3). Product Info Amount : 5 µg Purification : Greater than 85% as determined by SDS-PAGE. The PRKAB1 protein solution (0.5mg/ml) contains 20mM Tris-HCl buffer (pH 8.0), 0.15M NaCl, Content : 10% glycerol and 1mM DTT. Store at 4°C if entire vial will be used within 2-4 weeks. Store, frozen at -20°C for longer periods of Storage condition : time. -

Effects of Rapamycin on Social Interaction Deficits and Gene
Kotajima-Murakami et al. Molecular Brain (2019) 12:3 https://doi.org/10.1186/s13041-018-0423-2 RESEARCH Open Access Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero Hiroko Kotajima-Murakami1,2, Toshiyuki Kobayashi3, Hirofumi Kashii1,4, Atsushi Sato1,5, Yoko Hagino1, Miho Tanaka1,6, Yasumasa Nishito7, Yukio Takamatsu7, Shigeo Uchino1,2 and Kazutaka Ikeda1* Abstract The mammalian target of rapamycin (mTOR) signaling pathway plays a crucial role in cell metabolism, growth, and proliferation. The overactivation of mTOR has been implicated in the pathogenesis of syndromic autism spectrum disorder (ASD), such as tuberous sclerosis complex (TSC). Treatment with the mTOR inhibitor rapamycin improved social interaction deficits in mouse models of TSC. Prenatal exposure to valproic acid (VPA) increases the incidence of ASD. Rodent pups that are exposed to VPA in utero have been used as an animal model of ASD. Activation of the mTOR signaling pathway was recently observed in rodents that were exposed to VPA in utero, and rapamycin ameliorated social interaction deficits. The present study investigated the effect of rapamycin on social interaction deficits in both adolescence and adulthood, and gene expressions in mice that were exposed to VPA in utero. We subcutaneously injected 600 mg/kg VPA in pregnant mice on gestational day 12.5 and used the pups as a model of ASD. The pups were intraperitoneally injected with rapamycin or an equal volume of vehicle once daily for 2 consecutive days. The social interaction test was conducted in the offspring after the last rapamycin administration at 5–6 weeks of ages (adolescence) or 10–11 weeks of age (adulthood). -

Human Kinome Profiling Identifies a Requirement for AMP-Activated
Human kinome profiling identifies a requirement for AMP-activated protein kinase during human cytomegalovirus infection Laura J. Terrya, Livia Vastagb,1, Joshua D. Rabinowitzb, and Thomas Shenka,2 aDepartment of Molecular Biology and bDepartment of Chemistry and the Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NJ 08544 Contributed by Thomas Shenk, January 11, 2012 (sent for review December 29, 2011) Human cytomegalovirus (HCMV) modulates numerous cellular (7). Thus, the connections between AMPK activity and metabolic signaling pathways. Alterations in signaling are evident from the changes during HCMV infection have remained unclear. broad changes in cellular phosphorylation that occur during HCMV We confirmed the requirement for AMPK during infection, infection and from the altered activity of multiple kinases. Here we and we show that an AMPK antagonist, compound C, blocks report a comprehensive RNAi screen, which predicts that 106 cellular HCMV-induced changes to glycolysis and inhibits viral gene kinases influence growth of the virus, most of which were not expression. These studies argue that AMPK or a related, com- previously linked to HCMV replication. Multiple elements of the pound C-sensitive kinase is an essential contributor to metabolic AMP-activated protein kinase (AMPK) pathway scored in the screen. changes initiated by HCMV and provide unique insight into As a regulator of carbon and nucleotide metabolism, AMPK is poised potential antiviral strategies. to activate many of the metabolic pathways induced by HCMV infection. An AMPK inhibitor, compound C, blocked a substantial Results portion of HCMV-induced metabolic changes, inhibited the accumu- HumanKinomeScreenIdentifies Putative Effectors of HCMV Replication. lation of all HCMV proteins tested, and markedly reduced the We conducted an siRNA screen of the human kinome to perform an production of infectious progeny. -

A New Computational Approach to Evaluating Systemic Gene–Gene Interactions in a Pathway Affected by Drug LY294002
processes Article A New Computational Approach to Evaluating Systemic Gene–Gene Interactions in a Pathway Affected by Drug LY294002 Shinuk Kim College of Kyedang General Education, Sangmyung University, Cheonan 31066, Korea; [email protected]; Tel.: +82-(41)-550-5452; Fax: +82-(41)-550-5439 Received: 14 August 2020; Accepted: 23 September 2020; Published: 1 October 2020 Abstract: In this study, we investigate how drugs systemically affect genes via pathways by integrating information from interactions between chemical compounds and molecular expression datasets, and from pathway information such as gene sets using mathematical models. First, we adopt drug-induced gene expression datasets; then, employ gene set enrichment analysis tools for selecting candidate enrichment pathways; and lastly, implement the inverse algorithm package for identifying gene–gene regulatory networks in a pathway. We tested LY294002-induced datasets of the MCF7 breast cancer cell lines, and found a CELL CYCLE pathway with 101 genes, ERBB signaling pathway consisting of 82 genes, and MTOR pathway consisting of 45 genes. We consider two interactions: quantity strength depending on number of interactions, and quality strength depending on weight of interaction as positive (+) and negative ( ) interactions. Our methods revealed ANAPC1-CDK6 ( 0.412) and − − ORC2L- CHEK1(0.951) for the CELL CYCLE pathway; INS-RPS6 ( 3.125) and PRKAA2-PRKAA2 − (+1.319) for the MTOR pathway; and CBLB-RPS6KB1 ( 0.141), RPS6KB1-CBLC (+0.238) for the ERBB − signaling pathway to be top quality interactions. Top quantity interactions discovered include 12; the CDC ( ,+) gene family for the CELL CYCLE pathway, 20; PIK3 ( ), 23; PIK3CG (+) for the MTOR − − pathway, 11; PAK ( ), 10; PIK3 (+) for the ERBB signaling pathway. -

NFKBIZ Mutation Prevalence in the Arthur/Schmitz/Chapuy Cohorts
Supplemental Tables/Figures: Table S1: NFKBIZ mutation prevalence in the Arthur/Schmitz/Chapuy cohorts. Subtype Cohort All ABC DLC Schmitz Chapuy # patients 1006 511 330 466 210 UTR 101 (10) 66 (13) 39 (12) 49 (11) 13 (6) Amplification 86 (8.5) 66 (12.9) 23 (7) 35 (8) 28 (13) NFKBIZ Mutation TOTAL 174 (17) 120 (23.5) 57 (17) 79 (17) 38 (18) Table S2: NFKBIZ 3′ UTR mutations in other WGS cohorts. Cohort # Patients NFKBIZ UTR mutations (%) Publication BL_Adult 81 1 (1.2) Grande_et_al,2019 BL_Pediatric 124 1 (0.8) unpublished DLBCL_cell_lines 15 2 (13.3) Morin_et_al,2013 DLBCL_BC 117 11 (9.4) Arthur_et_al,2018 DLBCL_ICGC 87 11 (12.6) Hübschmann_et_al,2021 FL_ICGC 100 4 (4) Hübschmann_et_al,2021 FL_Kridel 48 1 (2.1) Kridel_et_al,2016 Table S3: COO and LymphGen classifications of DLBCLs and FLs from other cohorts. NFKBIZ UTR mutations (%) DLBCL (%) FL (%) ABC 13 (41.9) 13 (54.2) 0 (0) GCB 4 (12.9) 4 (16.7) 0 (0) COO UNCLASS 2 (6.5) 2 (8.3) 0 (0) NA 12 (38.7) 5 (20.8) 5 (100) TOTAL 31 24 5 BN2 13 (41.9) 9 (37.5) 3 (60) EZB 4 (12.9) 2 (8.3) 2 (40) ST2 1 (3.2) 1 (4.2) 0 (0) LymphGen Other 9 (29) 8 (33.3) 0 (0) Composite 2 (6.5) 2 (8.3) 0 (0) NA 2 (6.5) 2 (8.3) 0 (0) TOTAL 31 24 5 Table S4: LymphGen classifications of all patients and those with NFKBIZ mutations in Arthur/Schmitz/Chapuy cohorts. -

Association Study of AMP-Activated Protein Kinase Subunit Genes In
European Journal of Endocrinology (2009) 161 405–409 ISSN 0804-4643 CLINICAL STUDY Association study of AMP-activated protein kinase subunit genes in polycystic ovary syndrome Kari Sproul1,2, Michelle R Jones3, Ricardo Azziz1,2,4 and Mark O Goodarzi1,3,4,5 1Department of Obstetrics and Gynecology, Cedars-Sinai Medical Center, Los Angeles, California 90048, USA, 2Department of Obstetrics and Gynecology, the David Geffen School of Medicine at UCLA, Los Angeles, California 90095, USA, 3Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, Cedars-Sinai Medical Center, 8700 Beverly Boulevard, Room B-131, Los Angeles, California 90048, USA, 4Department of Medicine, the David Geffen School of Medicine at UCLA, Los Angeles, California 90095, USA and 5Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, California 90048, USA (Correspondence should be addressed to M O Goodarzi at Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, Cedars-Sinai Medical Center; Email: [email protected]) Abstract Objective: To examine the genes for AMP-activated protein kinase (AMPK) subunits a2(PRKAA2) and g3(PRKAG3) as candidates for polycystic ovary syndrome (PCOS) and its component traits. Design and methods: A total of 287 white PCOS women were recruited from the reproductive endocrinology clinic at the University of Alabama at Birmingham and 187 white control subjects were recruited from the surrounding community. Seven PRKAA2 single nucleotide polymorphisms (SNPs) and four PRKAG3 SNPs were genotyped in PCOS cases and controls. Genotyping and association analysis were performed at Cedars-Sinai Medical Center. Results: Nominal associations of PRKAA2 variants with insulin-related traits and the PRKAG3 Pro71Ala variant with PCOS were not statistically significant after multiple testing correction. -
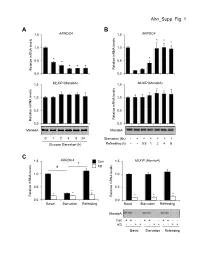
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

Phosphoproteomics of Retinoblastoma: a Pilot Study Identifies Aberrant Kinases
molecules Article Phosphoproteomics of Retinoblastoma: A Pilot Study Identifies Aberrant Kinases Lakshmi Dhevi Nagarajha Selvan 1,†, Ravikanth Danda 1,2,†, Anil K. Madugundu 3 ID , Vinuth N. Puttamallesh 3, Gajanan J. Sathe 3,4, Uma Maheswari Krishnan 2, Vikas Khetan 5, Pukhraj Rishi 5, Thottethodi Subrahmanya Keshava Prasad 3,6 ID , Akhilesh Pandey 3,7,8, Subramanian Krishnakumar 1, Harsha Gowda 3,* and Sailaja V. Elchuri 9,* 1 L&T Opthalmic Pathology, Vision Research Foundation, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India; [email protected] (L.D.N.S.); [email protected] (R.D.); [email protected] (S.K.) 2 Centre for Nanotechnology and Advanced Biomaterials, Shanmugha Arts, Science, Technology and Research Academy University, Tanjore, Tamil Nadu 613 401, India; [email protected] 3 Institute of Bioinformatics, International Technology Park, Bangalore, Karnataka 560 066, India; [email protected] (A.K.M.); [email protected] (V.N.P.); [email protected] (G.J.S.); [email protected] (T.S.K.P.); [email protected] (A.P.) 4 Manipal Academy of Higher Education (MAHE), Manipal, Karnataka 576 104, India 5 Shri Bhagwan Mahavir Vitreoretinal Services, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India; [email protected] (V.K.); [email protected] (P.R.) 6 Center for Systems Biology and Molecular Medicine, Yenepoya Research Centre, Yenepoya (Deemed to be University), Mangalore, Karnataka 575 108, India 7 McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 8 Departments of Biological Chemistry, Pathology and Oncology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 9 Department of Nanotechnology, Vision Research Foundation, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India * Correspondence: [email protected] (H.G.); [email protected] (S.V.E.); Tel.: +91-80-28416140 (H.G.); +91-44-28271616 (S.V.E.) † These authors contributed equally to this work.