A Phenotype for Enigmatic DNA Polymerase II: a Pivotal Role for Pol II in Replication Restart in UV-Irradiated Escherichia Coli
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
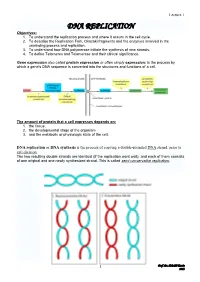
DNA REPLICATION Objectives: 1
Lecture 1 DNA REPLICATION Objectives: 1. To understand the replication process and where it occurs in the cell cycle. 2. To describe the Replication Fork, Okazaki fragments and the enzymes involved in the unwinding process and replication. 3. To understand how DNA polymerase initiate the synthesis of new strands. 4. To define Telomeres and Telomerase and their clinical significance. Gene expression also called protein expression or often simply expression: is the process by which a gene's DNA sequence is converted into the structures and functions of a cell. The amount of protein that a cell expresses depends on: 1. the tissue, 2. the developmental stage of the organism 3. and the metabolic or physiologic state of the cell. DNA replication or DNA synthesis is the process of copying a double-stranded DNA strand, prior to cell division. The two resulting double strands are identical (if the replication went well), and each of them consists of one original and one newly synthesized strand. This is called semi conservative replication. 1 Prof. Dr. H.D.El-Yassin 2013 Lecture 1 The process of replication consists of three steps, initiation, replication and termination. 1. Prokaryotic replication Basic Requirement for DNA Synthesis 1. Substrates: the four deoxy nucleosides triphosphates are needed as substrates for DNA synthesis. Cleavage of the high-energy phosphate bond between the α and β phosphates provides the energy for the addition of the nucleotide. 2. Template: DNA replication cannot occur without a template. A template is required to direct the addition of the appropriate complementary deoxynucleotide to the newly synthesized DNA strand. -
DNA Polymerase Exchange and Lesion Bypass in Escherichia Coli
DNA Polymerase Exchange and Lesion Bypass in Escherichia Coli The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Kath, James Evon. 2016. DNA Polymerase Exchange and Lesion Bypass in Escherichia Coli. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26718716 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ! ! ! ! ! ! ! DNA!polymerase!exchange!and!lesion!bypass!in!Escherichia)coli! ! A!dissertation!presented! by! James!Evon!Kath! to! The!Committee!on!Higher!Degrees!in!Biophysics! ! in!partial!fulfillment!of!the!requirements! for!the!degree!of! Doctor!of!Philosophy! in!the!subject!of! Biophysics! ! Harvard!University! Cambridge,!Massachusetts! October!2015! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ©!2015!L!James!E.!Kath.!Some!Rights!Reserved.! ! This!work!is!licensed!under!the!Creative!Commons!Attribution!3.0!United!States!License.!To! view!a!copy!of!this!license,!visit:!http://creativecommons.org/licenses/By/3.0/us! ! ! Dissertation!Advisor:!Professor!Joseph!J.!Loparo! ! ! !!!!!!!!James!Evon!Kath! ! DNA$polymerase$exchange$and$lesion$bypass$in$Escherichia)coli$ $ Abstract$ ! Translesion! synthesis! (TLS)! alleviates! -

Conversion of OX174 and Fd Single-Stranded DNA to Replicative Forms in Extracts of Escherichia Coli (Dnac, Dnad, and Dnag Gene Products/DNA Polymerase III) REED B
Proc. Nat. Acad. Sci. USA Vol. 69, No. 11, pp. 3233-3237, November 1972 Conversion of OX174 and fd Single-Stranded DNA to Replicative Forms in Extracts of Escherichia coli (dnaC, dnaD, and dnaG gene products/DNA polymerase III) REED B. WICKNER, MICHEL WRIGHT, SUE WICKNER, AND JERARD HURWITZ Department of Developmental Biology and Cancer, Division of Biological Sciences, Albert Einstein College of Medicine, Bronx, New York 10461 Communicated by Harry Eagle, August 28, 1972 ABSTRACT 4X174 and M13 (fd) single-stranded cir- MATERIALS AND METHODS cular DNAs are converted to their replicative forms by ex- tracts of E. coli pol Al cells. We find that the qX174 DNA- [a-32P]dTTP was obtained from New England Nuclear Corp. dependent reaction requires Mg++, ATP, and all four de- OX174 DNA was prepared by the method of Sinsheimer (7) oxynucleoside triphosphates, but not CTP, UTP, or GTP. or Franke and Ray (8), while fd viral DNA was prepared as This reaction also involves the products of the dnaC, dnaD, dnaE (DNA polymerase III), and dnaG genes, described (9). Pancreatic RNase was the highest grade ob- but not that of dnaF (ribonucleotide reductase). The in tainable from Worthington Biochemical Corp. It was further vitro conversion of fd single-stranded DNA to the replica- freed of possible contaminating DNase by heating a solution tive form requires all four ribonucleoside triphosphates, (2 mg/ml in 15 mM sodium citrate, pH 5) at 800 for 10 min. Mg++, and all four deoxynucleoside triphosphates. The E. protein was purified from E. coli strain reaction involves the product of gene dnaE but not those coli unwinding of genes dnaC, dnaD, dnaF, or dnaG. -

Product Sheet Info
Master Clone List for NR-19279 ® Vibrio cholerae Gateway Clone Set, Recombinant in Escherichia coli, Plates 1-46 Catalog No. NR-19279 Table 1: Vibrio cholerae Gateway® Clones, Plate 1 (NR-19679) Clone ID Well ORF Locus ID Symbol Product Accession Position Length Number 174071 A02 367 VC2271 ribD riboflavin-specific deaminase NP_231902.1 174346 A03 336 VC1877 lpxK tetraacyldisaccharide 4`-kinase NP_231511.1 174354 A04 342 VC0953 holA DNA polymerase III, delta subunit NP_230600.1 174115 A05 388 VC2085 sucC succinyl-CoA synthase, beta subunit NP_231717.1 174310 A06 506 VC2400 murC UDP-N-acetylmuramate--alanine ligase NP_232030.1 174523 A07 132 VC0644 rbfA ribosome-binding factor A NP_230293.2 174632 A08 322 VC0681 ribF riboflavin kinase-FMN adenylyltransferase NP_230330.1 174930 A09 433 VC0720 phoR histidine protein kinase PhoR NP_230369.1 174953 A10 206 VC1178 conserved hypothetical protein NP_230823.1 174976 A11 213 VC2358 hypothetical protein NP_231988.1 174898 A12 369 VC0154 trmA tRNA (uracil-5-)-methyltransferase NP_229811.1 174059 B01 73 VC2098 hypothetical protein NP_231730.1 174075 B02 82 VC0561 rpsP ribosomal protein S16 NP_230212.1 174087 B03 378 VC1843 cydB-1 cytochrome d ubiquinol oxidase, subunit II NP_231477.1 174099 B04 383 VC1798 eha eha protein NP_231433.1 174294 B05 494 VC0763 GTP-binding protein NP_230412.1 174311 B06 314 VC2183 prsA ribose-phosphate pyrophosphokinase NP_231814.1 174603 B07 108 VC0675 thyA thymidylate synthase NP_230324.1 174474 B08 466 VC1297 asnS asparaginyl-tRNA synthetase NP_230942.2 174933 B09 198 -

Roles of DNA Polymerases V and II in SOS-Induced Error-Prone and Error-Free Repair in Escherichia Coli
Colloquium Roles of DNA polymerases V and II in SOS-induced error-prone and error-free repair in Escherichia coli Phuong Pham*, Savithri Rangarajan*, Roger Woodgate†, and Myron F. Goodman*‡ *Departments of Biological Sciences and Chemistry, Hedco Molecular Biology Laboratories, University of Southern California, Los Angeles, CA 90089-1340; and †Section on DNA Replication, Repair and Mutagenesis, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892-2725 DNA polymerase V, composed of a heterotrimer of the DNA regulated at the transcriptional level by the LexA protein (14). damage-inducible UmuC and UmuD2 proteins, working in conjunc- Many of these genes encode proteins required to repair DNA tion with RecA, single-stranded DNA (ssDNA)-binding protein damage (15). The overriding importance of DNA repair is (SSB),  sliding clamp, and ␥ clamp loading complex, are respon- apparent from the observation that a single pyrimidine dimer is sible for most SOS lesion-targeted mutations in Escherichia coli,by lethal in E. coli strains defective for excision and recombinational catalyzing translesion synthesis (TLS). DNA polymerase II, the repair (16). These experiments were among the first to demon- product of the damage-inducible polB (dinA ) gene plays a pivotal strate the essential contribution of DNA repair to cell survival. role in replication-restart, a process that bypasses DNA damage in Excision and recombination repair pathways are referred to as an error-free manner. Replication-restart takes place almost im- ‘‘error-free’’ because they do not result in an increase in muta- mediately after the DNA is damaged (Ϸ2 min post-UV irradiation), tion rate above spontaneous background levels (1). -

DNA Polymerase I- Dependent Replication (Temperature-Sensitive Dna Mutants/Extragenic Suppression) OSAMI NIWA*, SHARON K
Proc. Nati Acad. Sci. USA Vol. 78, No. 11, pp. 7024-7027, November 1981 Genetics Alternate pathways of DNA replication: DNA polymerase I- dependent replication (temperature-sensitive dna mutants/extragenic suppression) OSAMI NIWA*, SHARON K. BRYAN, AND ROBB E. MOSES Department ofCell Biology, Baylor College of Medicine, Houston, Texas 77030 Communicated by D. Nathans, July 10, 1981 ABSTRACT We have previously shown that someEscherichia proceed in the presence of a functional DNA polymerase I ac- coli [derivatives of strain HS432 (polAl, polB100, polC1026)] can tivity, despite a ts DNA polymerase III (6). replicate DNA at a restrictive temperature in the presence of a We report here that DNA replication in the parent strain polCts mutation and that such revertants contain apparent DNA becomes temperature-resistant with introduction ofDNA poly- polymerase I activity. We demonstrate here that this strain ofE. merase I activity but is ts in the absence of DNA polymerase colibecomes temperature-resistant upon the introduction ofa nor- I or presence of a ts DNA polymerase I activity. We conclude mal gene for DNA polymerase I or suppression of the polAl non- that this strain contains a sense mutation. Such temperature-resistant phenocopies become mutation (pcbA-) that allows repli- temperature-sensitive upon introduction of a temperature-sensi- cation to be dependent on DNA polymerase I polymerizing tive DNA polymerase I gene. Our results confirm that DNA rep- activity. This locus can be transduced to other E. coli strains and lication is DNA polymerase I-dependent in the temperature-re- again exerts phenotypic suppression of the polCts mutation in sistant revertants, indicating that an alternative pathway of the presence of DNA polymerase I. -

Biotechnology DNA Polymerases
Paper No. : 04 Genetic engineering and recombinant DNA technology Module : 07 DNA polymerases Principal Investigator: Dr Vibha Dhawan, Distinguished Fellow and Sr. Director The Energy and Resouurces Institute (TERI), New Delhi Co-Principal Investigator: Prof S K Jain, Professor, of Medical Biochemistry Jamia Hamdard University, New Delhi Paper Coordinator: Dr Mohan Chandra Joshi, Assistant Professor, Jamia Millia Islamia, New Delhi Content Writer: Dr Samer Singh, Assistant Professor, Panjab University, Chandigarh Content Reviwer: Dr Mohan Chandra Joshi, Assistant Professor, Jamia Millia Islamia, New Delhi Genetic engineering and recombinant DNA technology Biotechnology DNA polymerases Description of Module Subject Name Biotechnology Paper Name Genetic engineering and recombinant DNA technology Module Name/Title DNA polymerases Module Id 07 Pre-requisites Basic DNA Structure and DNA replication Objectives 1) WHAT ARE DNA POLYMERASES? - DISCOVERY 2) TYPES in Escherichia coli 3) WHAT THEY DO? 4) STRUCTURE - CONSERVATION & FUNCTION 5) DNA POLYMERIZATION REACTION and ITS ATTRIBUTES 6) DNA POLYMERASE'S FIDELITY AND PROCESSIVITY 7) DNA POLYMERASE FAMILY AND EXAMPLES 8) HOW SPECIAL FUNCTION DNA POLYMERASE 'TELOMERASE' CARRIES OUT LENGTHENING OF LINEAR DNA ENDS Keywords DNA polymerase, reverse transcriptase, DNA polymerization, Processivity, Fidelity, Telomerase Genetic engineering and recombinant DNA technology Biotechnology DNA polymerases Table of contents A. Learning Objectives B. Keywords C. DNA POLYMERASES C1. General Overview -Discovery C2. Comparison of DNA polymerases of Escherichia coli C3. DNA POLYMERASE CORE STRUCTURE & FUNCTION C4. DNA POLYMERIZATION ACTIVITY and ITS ATTRIBUTES C5. DNA POLYMERASE FAMILIES C6. TELOMERASE- A special function DNA polymerase in eukaryotes D. Summary A. LEARNING OBJECTIVES In this module, Students will learn following: 1. WHAT ARE DNA POLYMERASES? - DISCOVERY 2. -

Table 4. V. Cholerae Flexgene ORF Collection
Table 4. V. cholerae FLEXGene ORF collection Reference Clone protein PlasmID clone GenBank Locus tag Symbol accession identifier FLEX clone name accession Product name VC0001 NP_062585 VcCD00019918 FLH200476.01F DQ772770 hypothetical protein VC0002 mioC NP_062586 VcCD00019938 FLH200506.01F DQ772771 mioC protein VC0003 thdF NP_062587 VcCD00019958 FLH200531.01F DQ772772 thiophene and furan oxidation protein ThdF VC0004 yidC NP_062588 VcCD00019970 FLH200545.01F DQ772773 inner membrane protein, 60 kDa VC0005 NP_062589 VcCD00061243 FLH236482.01F DQ899316 conserved hypothetical protein VC0006 rnpA NP_062590 VcCD00025697 FLH214799.01F DQ772774 ribonuclease P protein component VC0007 rpmH NP_062591 VcCD00061229 FLH236450.01F DQ899317 ribosomal protein L34 VC0008 NP_062592 VcCD00019917 FLH200475.01F DQ772775 amino acid ABC transporter, ATP-binding protein VC0009 NP_062593 VcCD00019966 FLH200540.01F DQ772776 amino acid ABC transproter, permease protein VC0010 NP_062594 VcCD00019152 FLH199275.01F DQ772777 amino acid ABC transporter, periplasmic amino acid-binding portion VC0011 NP_062595 VcCD00019151 FLH199274.01F DQ772778 hypothetical protein VC0012 dnaA NP_062596 VcCD00017363 FLH174286.01F DQ772779 chromosomal DNA replication initiator DnaA VC0013 dnaN NP_062597 VcCD00017316 FLH174063.01F DQ772780 DNA polymerase III, beta chain VC0014 recF NP_062598 VcCD00019182 FLH199319.01F DQ772781 recF protein VC0015 gyrB NP_062599 VcCD00025458 FLH174642.01F DQ772782 DNA gyrase, subunit B VC0016 NP_229675 VcCD00019198 FLH199346.01F DQ772783 hypothetical protein -

The Escherichia Coli Polb Gene, Which Encodes DNA Polymerase II, Is Regulated by the SOS System HIROSHI IWASAKI,' ATSUO NAKATA,1 GRAHAM C
JOURNAL OF BACTERIOLOGY, Nov. 1990, p. 6268-6273 Vol. 172, No. 11 0021-9193/90/116268-06$02.00/0 Copyright © 1990, American Society for Microbiology The Escherichia coli polB Gene, Which Encodes DNA Polymerase II, Is Regulated by the SOS System HIROSHI IWASAKI,' ATSUO NAKATA,1 GRAHAM C. WALKER,2 AND HIDEO SHINAGAWA1* Department ofExperimental Chemotherapy, Research Institute for Microbial Disease, Osaka University, Suita, Osaka 565, Japan,1 and Biology Department, Massachusetts Institute of Technology, Cambridge, Massachusetts 021392 Received 7 June 1990/Accepted 13 August 1990 The dinA (damage inducible) gene was previously identified as one of the SOS genes with no known function; it was mapped near the leuB gene, where the poiB gene encoding DNA polymerase H was also mapped. We cloned the chromosomal fragment carrying the dinA region from the ordered Escherichia coli genomic library and mapped the dinA promoter precisely on the physical map of the chromosome. The cells that harbored multicopy plasmids with the dimA region expressed very high levels of DNA polymerase activity, which was sensitive to N-ethylmaleimide, an inhibitor of DNA polymerase II. Expression of the polymerase activity encoded by the dinA locus was regulated by SOS system, and the dinA promoter was the promoter of the gene encoding the DNA polymerase. From these data we conclude that the polB gene is identical to the dinA gene and is regulated by the SOS system. The product of the polB (dinA) gene was identified as an 80-kDa protein by the maxicell method. In Escherichia coli, three DNA polymerases have been pSY343 (27), pSCH18 (10), and pRS528 (22) were used for identified (15). -

Reconstitution of Repair-Gap 1V Mutagenesis with Purified Proteins from Escherichia Coli: a Role for DNA Polymerases III and II
Proc. Natl. Acad. Sci. USA Vol. 93, pp. 1376-1380, February 1996 Biochemistry Reconstitution of repair-gap 1V mutagenesis with purified proteins from Escherichia coli: A role for DNA polymerases III and II (DNA repair/excision repair/error-prone repair/carcinogenesis) GuY TOMER*, ORNA COHEN-FIX*, MICHAEL O'DONNELLt, MYRON GOODMANt, AND Zvi LIVNEH*§ *Department of Biochemistry, Weizmann Institute of Science, Rehovot 76100, Israel; tDepartment of Microbiology, Cornell University Medical College, New York, NY 10021; and tDepartment of Molecular Biology, University of Southern California, Los Angeles, CA 90089 Communicated by I. Robert Lehman, Stanford University Medical Center, Stanford, CA, October 26, 1995 ABSTRACT Using a cell-free system for UV mutagenesis, identified two pathways: type I, or replicative UV mutagenesis, we have previously demonstrated the existence of a mutagenic that depended on DNA replication, and type II, or repair-gap pathway associated with nucleotide-excision repair gaps. UV mutagenesis, that depended on nucleotide excision repair Here, we report that this pathway can be reconstituted by (15, 16). Here, we report the reconstitution and characteriza- using six purified proteins: UvrA, UvrB, UvrC, DNA helicase tion of repair-gap UV mutagenesis using purified components. II, DNA polymerase III core, and DNA ligase. This establishes the minimal requirements for repair-gap UV mutagenesis. DNA polymerase II could replace DNA polymerase III, al- MATERIALS AND METHODS though less effectively, whereas DNA polymerase I, the major Materials. The sources of materials were as follows: unla- repair polymerase, could not. DNA sequence analysis of beled dNTPs and creatine phosphate, Boehringer Mannheim; mutations generated in the in vitro reaction revealed a spec- [a-32P]dNTPs (400 Ci/mmol; 1 Ci = 37 GBq), Amersham; trum typical of mutations targeted to UV lesions. -

NAPRALERT Classification Codes
NAPRALERT Classification Codes June 1993 STN International® Copyright © 1993 American Chemical Society Quoting or copying of material from this publication for educational purposes is encouraged, providing acknowledgement is made of the source of such material. Classification Codes in NAPRALERT The NAPRALERT File contains classification codes that designate pharmacological activities. The code and a corresponding textual description are searchable in the /CC field. To be comprehensive, both the code and the text should be searched. Either may be posted, but not both. The following tables list the code and the text for the various categories. The first two digits of the code describe the categories. Each table lists the category described by codes. The last table (starting on page 56) lists the Classification Codes alphabetically. The text is followed by the code that also describes the category. General types of pharmacological activities may encompass several different categories of effect. You may want to search several classification codes, depending upon how general or specific you want the retrievals to be. By reading through the list, you may find several categories related to the information of interest to you. For example, if you are looking for information on diabetes, you might want to included both HYPOGLYCEMIC ACTIVITY/CC and ANTIHYPERGLYCEMIC ACTIVITY/CC and their codes in the search profile. Use the EXPAND command to verify search terms. => S HYPOGLYCEMIC ACTIVITY/CC OR 17006/CC OR ANTIHYPERGLYCEMIC ACTIVITY/CC OR 17007/CC 490 “HYPOGLYCEMIC”/CC 26131 “ACTIVITY”/CC 490 HYPOGLYCEMIC ACTIVITY/CC ((“HYPOGLYCEMIC”(S)”ACTIVITY”)/CC) 6 17006/CC 776 “ANTIHYPERGLYCEMIC”/CC 26131 “ACTIVITY”/CC 776 ANTIHYPERGLYCEMIC ACTIVITY/CC ((“ANTIHYPERGLYCEMIC”(S)”ACTIVITY”)/CC) 3 17007/CC L1 1038 HYPOGLYCEMIC ACTIVITY/CC OR 17006/CC OR ANTIHYPERGLYCEMIC ACTIVITY/CC OR 17007/CC 2 This search retrieves records with the searched classification codes such as the ones shown here. -

MEMORIAL SLOAN-KETTERING CANCER CENTER Curriculum
MEMORIAL SLOAN-KETTERING CANCER CENTER Curriculum Vitae and Bibliography Name: Jerard Hurwitz Date of Birth: November 20, 1928 Place of Birth: New York, New York Nationality: U.S. Citizen Office Address: Memorial Sloan-Kettering Cancer Center Telephone #: 212-794-5896 1275 York Avenue E-Mail: [email protected] New York, NY 10021 Fax#: (212) 717-3627 Home Address: 418 East 59th Street Telephone No. 212-223-4364 New York, NY 10022 Education: Indiana University 1949, B.A., Chemistry Western Reserve University 1953, Ph.D., Biochemistry Postdoctoral Training: 1953-1954 American Cancer Society, Postdoctoral Fellow National Medical Research Council, Mill Hill, London, England 1955-1956 American Cancer Society, Postdoctoral Fellow National Institutes of Health, Bethesda, Maryland Positions and Appointments: 1956-1958 Instructor of Microbiology, Washington University, St. Louis, MO 1958-1960 Assistant Professor of Microbiology, New York University School of Medicine, New York, New York 1960-1963 Associate Professor of Microbiology, New York University School of Medicine, New York, New York 1963-1965 Professor of Molecular Biology, Albert Einstein College of Medicine, Bronx, New York 1965-1984 Professor and Chairman Dept. of Developmental Biology and Cancer, Albert Einstein College of Medicine, Bronx, New York 1984-Present Member, Sloan-Kettering Institute Professor, Sloan-Kettering Division of the Cornell University Medical College, New York, New York 1989-Present Head, William Randolph Hearst Laboratory of Radiation Biology, Memorial Sloan-Kettering Cancer Center, New York, New York 1991-2003 Vice-chairman, Sloan-Kettering Institute, Memorial Sloan-Kettering Cancer Center, New York, New York Scientific and Medical Societies: American Society of Biological Chemists New York Academy of Sciences Member of the National Academy of Sciences American Academy of Arts and Sciences Honors and Awards: 1962 Eli Lilly Award in Biochemistry 1962 Sigma Xi Award 1963-1998 American Cancer Society Professorship 1964-1968 N000.I.H.