Mutational Analysis of the P. Falciparum ARO Protein, Functional Analysis of Its Predicted Binding Partner AIP and Identification Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Basal Body Structure and Composition in the Apicomplexans Toxoplasma and Plasmodium Maria E
Francia et al. Cilia (2016) 5:3 DOI 10.1186/s13630-016-0025-5 Cilia REVIEW Open Access Basal body structure and composition in the apicomplexans Toxoplasma and Plasmodium Maria E. Francia1* , Jean‑Francois Dubremetz2 and Naomi S. Morrissette3 Abstract The phylum Apicomplexa encompasses numerous important human and animal disease-causing parasites, includ‑ ing the Plasmodium species, and Toxoplasma gondii, causative agents of malaria and toxoplasmosis, respectively. Apicomplexans proliferate by asexual replication and can also undergo sexual recombination. Most life cycle stages of the parasite lack flagella; these structures only appear on male gametes. Although male gametes (microgametes) assemble a typical 9 2 axoneme, the structure of the templating basal body is poorly defined. Moreover, the rela‑ tionship between asexual+ stage centrioles and microgamete basal bodies remains unclear. While asexual stages of Plasmodium lack defined centriole structures, the asexual stages of Toxoplasma and closely related coccidian api‑ complexans contain centrioles that consist of nine singlet microtubules and a central tubule. There are relatively few ultra-structural images of Toxoplasma microgametes, which only develop in cat intestinal epithelium. Only a subset of these include sections through the basal body: to date, none have unambiguously captured organization of the basal body structure. Moreover, it is unclear whether this basal body is derived from pre-existing asexual stage centrioles or is synthesized de novo. Basal bodies in Plasmodium microgametes are thought to be synthesized de novo, and their assembly remains ill-defined. Apicomplexan genomes harbor genes encoding δ- and ε-tubulin homologs, potentially enabling these parasites to assemble a typical triplet basal body structure. -
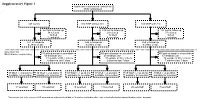
Supplementary Figure 1 Multicenter Randomised Control Trial 2746 Randomised
Supplementary Figure 1 Multicenter randomised control trial 2746 randomised 947 control 910 MNP without zinc 889 MNP with zinc 223 lost to follow up 219 lost to follow up 183 lost to follow up 34 refused 29 refused 37 refused 16 died 12 died 9 died 3 excluded 4 excluded 1 excluded 671 in follow-up 646 in follow-up 659 in follow-up at 24mo of age at 24mo of age at 24mo of age Selection for Microbiome sequencing 516 paired samples unavailable 469 paired samples unavailable 497 paired samples unavailable 69 antibiotic use 63 antibiotic use 67 antibiotic use 31 outside of WLZ criteria 37 outside of WLZ criteria 34 outside of WLZ criteria 6 diarrhea last 7 days 2 diarrhea last 7 days 7 diarrhea last 7 days 39 WLZ > -1 at 24 mo 10 WLZ < -2 at 24mo 58 WLZ > -1 at 24 mo 17 WLZ < -2 at 24mo 48 WLZ > -1 at 24 mo 8 WLZ < -2 at 24mo available for selection available for selection available for selection available for selection available for selection1 available for selection1 14 selected 10 selected 15 selected 14 selected 20 selected1 7 selected1 1 Two subjects (one in the reference WLZ group and one undernourished) had, at 12 months, no diarrhea within 1 day of stool collection but reported diarrhea within 7 days prior. Length, cm kg Weight, Supplementary Figure 2. Length (left) and weight (right) z-scores of children recruited into clinical trial NCT00705445 during the first 24 months of life. Median and quantile values are shown, with medians for participants profiled in current study indicated by red (undernourished) and black (reference WLZ) lines. -

Why the –Omic Future of Apicomplexa Should Include Gregarines Julie Boisard, Isabelle Florent
Why the –omic future of Apicomplexa should include Gregarines Julie Boisard, Isabelle Florent To cite this version: Julie Boisard, Isabelle Florent. Why the –omic future of Apicomplexa should include Gregarines. Biology of the Cell, Wiley, 2020, 10.1111/boc.202000006. hal-02553206 HAL Id: hal-02553206 https://hal.archives-ouvertes.fr/hal-02553206 Submitted on 24 Apr 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Article title: Why the –omic future of Apicomplexa should include Gregarines. Names of authors: Julie BOISARD1,2 and Isabelle FLORENT1 Authors affiliations: 1. Molécules de Communication et Adaptation des Microorganismes (MCAM, UMR 7245), Département Adaptations du Vivant (AVIV), Muséum National d’Histoire Naturelle, CNRS, CP52, 57 rue Cuvier 75231 Paris Cedex 05, France. 2. Structure et instabilité des génomes (STRING UMR 7196 CNRS / INSERM U1154), Département Adaptations du vivant (AVIV), Muséum National d'Histoire Naturelle, CP 26, 57 rue Cuvier 75231 Paris Cedex 05, France. Short Title: Gregarines –omics for Apicomplexa studies -

Tracing the Origin of Planktonic Protists in an Ancient Lake
microorganisms Article Tracing the Origin of Planktonic Protists in an Ancient Lake Nataliia V. Annenkova 1,* , Caterina R. Giner 2,3 and Ramiro Logares 2,* 1 Limnological Institute Siberian Branch of the Russian Academy of Sciences 3, Ulan-Batorskaya St., 664033 Irkutsk, Russia 2 Institute of Marine Sciences (ICM), CSIC, Passeig Marítim de la Barceloneta, 37-49, ES08003 Barcelona, Spain; [email protected] 3 Institute for the Oceans and Fisheries, University of British Columbia, 2202 Main Mall, Vancouver, BC V6T 1Z4, Canada * Correspondence: [email protected] (N.V.A.); [email protected] (R.L.) Received: 26 February 2020; Accepted: 7 April 2020; Published: 9 April 2020 Abstract: Ancient lakes are among the most interesting models for evolution studies because their biodiversity is the result of a complex combination of migration and speciation. Here, we investigate the origin of single celled planktonic eukaryotes from the oldest lake in the world—Lake Baikal (Russia). By using 18S rDNA metabarcoding, we recovered 1414 Operational Taxonomic Units (OTUs) belonging to protists populating surface waters (1–50 m) and representing pico/nano-sized cells. The recovered communities resembled other lacustrine freshwater assemblages found elsewhere, especially the taxonomically unclassified protists. However, our results suggest that a fraction of Baikal protists could belong to glacial relicts and have close relationships with marine/brackish species. Moreover, our results suggest that rapid radiation may have occurred among some protist taxa, partially mirroring what was already shown for multicellular organisms in Lake Baikal. We found 16% of the OTUs belonging to potential species flocks in Stramenopiles, Alveolata, Opisthokonta, Archaeplastida, Rhizaria, and Hacrobia. -

Assessing the Efficiency of Molecular Markers for the Species Identification of Gregarines Isolated from the Mealworm and Super Worm Midgut
Microorganisms 2018, 6, 119 1 of 20 Article Assessing the Efficiency of Molecular Markers for the Species Identification of Gregarines Isolated from the Mealworm and Super Worm Midgut Chiara Nocciolini 1, Claudio Cucini 2, Chiara Leo 2, Valeria Francardi 3, Elena Dreassi 4 and Antonio Carapelli 2,* 1 Polo d’Innovazione di Genomica Genetica e Biologia, 53100 Siena, Italy; [email protected] 2 Department of Life Sciences, University of Siena, 53100 Siena, Italy; [email protected] (C.C.); [email protected] (C.L.) 3 Consiglio per la ricerca in agricoltura e analisi dell’economia agraria – Centro di Difesa e Certificazione CREA-DC) Research Centre for Plant Protection and Certification, via di Lanciola 12/A, 50125 Firenze, Italy; [email protected] 4 Department of Biotechnology, Chemistry and Pharmacy, University of Siena, 53100 Siena, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-0577-234-410 Received: 28 September 2018; Accepted: 23 November 2018; Published: 27 November 2018 Abstract: Protozoa, of the taxon Gregarinasina, are a heterogeneous group of Apicomplexa that includes ~1600 species. They are parasites of a large variety of both marine and terrestrial invertebrates, mainly annelids, arthropods and mollusks. Unlike coccidians and heamosporidians, gregarines have not proven to have a negative effect on human welfare; thus, they have been poorly investigated. This study focuses on the molecular identification and phylogeny of the gregarine species found in the midgut of two insect species that are considered as an alternative source of animal proteins for the human diet: the mealworm Tenebrio molitor, and the super-worm Zophobas atratus (Coleoptera: Tenebrionidae). -

Gregarines (Apicomplexa: Gregarinasina) Are Monoxenous Parasites of Invertebrates
Gregarines (Apicomplexa: Gregarinasina) are monoxenous parasites of invertebrates. Those found in sand flies (Diptera: Psychodidae) and mosquitoes (Diptera: Culicidae) used to be considered a single eugregarine genus Ascogregarina. Our phylogenetic analyses of the gregarine SSU rDNA, including newly obtained sequences of three species from sand flies, showed that mosquito and sand fly gregarines are closely related to neogregarines, and most importantly, they form two disparate monophyletic groups. Based on these molecular features, accompanied by biological differences, we established a new genus Psychodiella for the gregarines from sand flies, reserving the genus Ascogregarina for the mosquito gregarines. In the new genus, two new species Psychodiella sergenti from Phlebotomus sergenti and Psychodiella tobbi from Phlebotomus tobbi were described. They differ in the life cycles (sexual development of Ps. sergenti is triggered by a blood meal intake) and morphology of their life stages, mainly oocysts. The susceptibility of five sand fly species to both gregarines showed their strict host specificity, as they were able to fully develop and complete the life cycle only in their natural hosts. The life cycle of Ps. sergenti was studied in detail using various microscopical methods. Oocysts are attached to the chorion of sand fly eggs. Sporozoites, with a three- layered pellicle and mucron, attach to the 1st instar larval intestine but are never located intracellularly. In the 4th instar larvae, the gregarines occur in the ectoperitrophic space and later in the intestinal lumen. In adults, the parasites appear in the body cavity, and the sexual development of Ps. sergenti takes place only in blood-fed females; gametocysts attach to the accessory glands and oocysts are injected into their lumen. -

Ellobiopsids of the Genus Thalassomyces Are Alveolates
J. Eukaryot. Microbiol., 51(2), 2004 pp. 246±252 q 2004 by the Society of Protozoologists Ellobiopsids of the Genus Thalassomyces are Alveolates JEFFREY D. SILBERMAN,a,b1 ALLEN G. COLLINS,c,2 LISA-ANN GERSHWIN,d,3 PATRICIA J. JOHNSONa and ANDREW J. ROGERe aDepartment of Microbiology, Immunology, and Molecular Genetics, University of California at Los Angeles, California, USA, and bInstitute of Geophysics and Planetary Physics, University of California at Los Angeles, California, USA, and cEcology, Behavior and Evolution Section, Division of Biology, University of California, La Jolla, California, USA, and dDepartment of Integrative Biology and Museum of Paleontology, University of California, Berkeley, California, USA, and eCanadian Institute for Advanced Research, Program in Evolutionary Biology, Genome Atlantic, Department of Biochemistry and Molecular Biology, Dalhousie University, Halifax, Nova Scotia, Canada ABSTRACT. Ellobiopsids are multinucleate protist parasites of aquatic crustaceans that possess a nutrient absorbing `root' inside the host and reproductive structures that protrude through the carapace. Ellobiopsids have variously been af®liated with fungi, `colorless algae', and dino¯agellates, although no morphological character has been identi®ed that de®nitively allies them with any particular eukaryotic lineage. The arrangement of the trailing and circumferential ¯agella of the rarely observed bi-¯agellated `zoospore' is reminiscent of dino¯agellate ¯agellation, but a well-organized `dinokaryotic nucleus' has never been observed. Using small subunit ribosomal RNA gene sequences from two species of Thalassomyces, phylogenetic analyses robustly place these ellobiopsid species among the alveolates (ciliates, apicomplexans, dino¯agellates and relatives) though without a clear af®liation to any established alveolate lineage. Our trees demonstrate that Thalassomyces fall within a dino¯agellate 1 apicomplexa 1 Perkinsidae 1 ``marine alveolate group 1'' clade, clustering most closely with dino¯agellates. -

Diversity of Extracellular Proteins During the Transition from the 'Proto
1 Diversity of extracellular proteins during the transition from the ‘proto-apicomplexan’ alveolates to the apicomplexan obligate parasites THOMAS J. TEMPLETON1,2* and ARNAB PAIN3,4 1 Department of Protozoology, Institute of Tropical Medicine (NEKKEN), Nagasaki University, 1-12-4 Sakamoto, Nagasaki 852-8523, Japan 2 Department of Microbiology and Immunology, Weill Cornell Medical College, New York 10021, USA 3 Pathogen Genomics Laboratory, Biological and Environmental Sciences and Engineering (BESE) Division, King Abdullah University of Science and Technology (KAUST), Thuwal, Jeddah 23955-6900, Kingdom of Saudi Arabia 4 Global Station for Zoonosis Control, Global Institution for Collaborative Research and Education (GI-CoRE), Hokkaido University, N20 W10 Kita-ku, Sapporo 001-0020, Japan (Received 22 May 2015; revised 12 August 2015; accepted 14 August 2015) SUMMARY The recent completion of high-coverage draft genome sequences for several alveolate protozoans – namely, the chromer- ids, Chromera velia and Vitrella brassicaformis; the perkinsid Perkinsus marinus; the apicomplexan, Gregarina niphandrodes, as well as high coverage transcriptome sequence information for several colpodellids, allows for new genome-scale com- parisons across a rich landscape of apicomplexans and other alveolates. Genome annotations can now be used to help in- terpret fine ultrastructure and cell biology, and guide new studies to describe a variety of alveolate life strategies, such as symbiosis or free living, predation, and obligate intracellular parasitism, as well to provide foundations to dissect the evo- lutionary transitions between these niches. This review focuses on the attempt to identify extracellular proteins which might mediate the physical interface of cell–cell interactions within the above life strategies, aided by annotation of the repertoires of predicted surface and secreted proteins encoded within alveolate genomes. -

Molecular Phylogeny and Surface Morphology of Colpodella Edax (Alveolata): Insights Into the Phagotrophic Ancestry of Apicomplexans
J. Eukaryot. Microbiol., 50(5), 2003 pp. 334±340 q 2003 by the Society of Protozoologists Molecular Phylogeny and Surface Morphology of Colpodella edax (Alveolata): Insights into the Phagotrophic Ancestry of Apicomplexans BRIAN S. LEANDER,a OLGA N. KUVARDINA,b VLADIMIR V. ALESHIN,b ALEXANDER P. MYLNIKOVc and PATRICK J. KEELINGa aCanadian Institute for Advanced Research, Program in Evolutionary Biology, Department of Botany, University of British Columbia, Vancouver, BC, V6T 1Z4, Canada, and bDepartments of Evolutionary Biochemistry and Invertebrate Zoology, Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, 119 992, Russian Federation, and cInstitute for the Biology of Inland Waters, Russian Academy of Sciences, Borok, Yaroslavskaya oblast, 152742, Russian Federation ABSTRACT. The molecular phylogeny of colpodellids provides a framework for inferences about the earliest stages in apicomplexan evolution and the characteristics of the last common ancestor of apicomplexans and dino¯agellates. We extended this research by presenting phylogenetic analyses of small subunit rRNA gene sequences from Colpodella edax and three unidenti®ed eukaryotes published from molecular phylogenetic surveys of anoxic environments. Phylogenetic analyses consistently showed C. edax and the environmental sequences nested within a colpodellid clade, which formed the sister group to (eu)apicomplexans. We also presented surface details of C. edax using scanning electron microscopy in order to supplement previous ultrastructural investigations of this species using transmission electron microscopy and to provide morphological context for interpreting environmental sequences. The microscopical data con®rmed a sparse distribution of micropores, an amphiesma consisting of small polygonal alveoli, ¯agellar hairs on the anterior ¯agellum, and a rostrum molded by the underlying (open-sided) conoid. -

Genetic Diversity and Habitats of Two Enigmatic Marine Alveolate Lineages
AQUATIC MICROBIAL ECOLOGY Vol. 42: 277–291, 2006 Published March 29 Aquat Microb Ecol Genetic diversity and habitats of two enigmatic marine alveolate lineages Agnès Groisillier1, Ramon Massana2, Klaus Valentin3, Daniel Vaulot1, Laure Guillou1,* 1Station Biologique, UMR 7144, CNRS, and Université Pierre & Marie Curie, BP74, 29682 Roscoff Cedex, France 2Department de Biologia Marina i Oceanografia, Institut de Ciències del Mar, CMIMA, CSIC. Passeig Marítim de la Barceloneta 37-49, 08003 Barcelona, Spain 3Alfred Wegener Institute for Polar Research, Am Handelshafen 12, 27570 Bremerhaven, Germany ABSTRACT: Systematic sequencing of environmental SSU rDNA genes amplified from different marine ecosystems has uncovered novel eukaryotic lineages, in particular within the alveolate and stramenopile radiations. The ecological and geographic distribution of 2 novel alveolate lineages (called Group I and II in previous papers) is inferred from the analysis of 62 different environmental clone libraries from freshwater and marine habitats. These 2 lineages have been, up to now, retrieved exclusively from marine ecosystems, including oceanic and coastal waters, sediments, hydrothermal vents, and perma- nent anoxic deep waters and usually represent the most abundant eukaryotic lineages in environmen- tal genetic libraries. While Group I is only composed of environmental sequences (118 clones), Group II contains, besides environmental sequences (158 clones), sequences from described genera (8) (Hema- todinium and Amoebophrya) that belong to the Syndiniales, an atypical order of dinoflagellates exclu- sively composed of marine parasites. This suggests that Group II could correspond to Syndiniales, al- though this should be confirmed in the future by examining the morphology of cells from Group II. Group II appears to be abundant in coastal and oceanic ecosystems, whereas permanent anoxic waters and hy- drothermal ecosystems are usually dominated by Group I. -

From Freshwater Fishes in Africa (Tomáš Scholz)
0 Organizer: Department of Botany and Zoology, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic Workshop venue: Instutute of Vertebrate Biology, Academy of Sciences CR Workshop date: 28 November 2018 Cover photo: Research on fish parasites throughout Africa: Fish collection in, Lake Turkana, Kenya; Fish examination in the Sudan; Teaching course on fish parasitology at the University of Khartoum, Sudan; Field laboratory in the Sudan Authors of cover photo: R. Blažek, A. de Chambrier and R. Kuchta All rights reserved. No part of this e-book may be reproduced or transmitted in any form or by any means without prior written permission of copyright administrator which can be contacted at Masaryk University Press, Žerotínovo náměstí 9, 601 77 Brno. © 2018 Masaryk University The stylistic revision of the publication has not been performed. The authors are fully responsible for the content correctness and layout of their contributions. ISBN 978-80-210-9079-8 ISBN 978-80-210-9083-5 (online: pdf) 1 Contents (We present only the first author in contents) ECIP Scientific Board ....................................................................................................................... 5 List of attendants ............................................................................................................................ 6 Programme ..................................................................................................................................... 7 Abstracts ........................................................................................................................................ -

Haemocystidium Spp., a Species Complex Infecting Ancient Aquatic
IDENTIFICACIÓN DE HEMOPARÁSITOS PRESENTES EN LA HERPETOFAUNA DE DIFERENTES DEPARTAMENTOS DE COLOMBIA. Leydy Paola González Camacho Universidad Nacional de Colombia Facultad de ciencias, Instituto de Biotecnología IBUN Bogotá, Colombia 2019 IDENTIFICACIÓN DE HEMOPARÁSITOS PRESENTES EN LA HERPETOFAUNA DE DIFERENTES DEPARTAMENTOS DE COLOMBIA. Leydy Paola González Camacho Tesis o trabajo de investigación presentada(o) como requisito parcial para optar al título de: Magister en Microbiología. Director (a): Ph.D MSc Nubia Estela Matta Camacho Codirector (a): Ph.D MSc Mario Vargas-Ramírez Línea de Investigación: Biología molecular de agentes infecciosos Grupo de Investigación: Caracterización inmunológica y genética Universidad Nacional de Colombia Facultad de ciencias, Instituto de biotecnología (IBUN) Bogotá, Colombia 2019 IV IDENTIFICACIÓN DE HEMOPARÁSITOS PRESENTES EN LA HERPETOFAUNA DE DIFERENTES DEPARTAMENTOS DE COLOMBIA. A mis padres, A mi familia, A mi hijo, inspiración en mi vida Agradecimientos Quiero agradecer especialmente a mis padres por su contribución en tiempo y recursos, así como su apoyo incondicional para la culminación de este proyecto. A mi hijo, Santiago Suárez, quien desde que llego a mi vida es mi mayor inspiración, y con quien hemos demostrado que todo lo podemos lograr; a Juan Suárez, quien me apoya, acompaña y no me ha dejado desfallecer, en este logro. A la Universidad Nacional de Colombia, departamento de biología y el posgrado en microbiología, por permitirme formarme profesionalmente; a Socorro Prieto, por su apoyo incondicional. Doy agradecimiento especial a mis tutores, la profesora Nubia Estela Matta y el profesor Mario Vargas-Ramírez, por el apoyo en el desarrollo de esta investigación, por su consejo y ayuda significativa con esta investigación.