A New Framework for the Study of Apicomplexan Diversity Across Environments
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Basal Body Structure and Composition in the Apicomplexans Toxoplasma and Plasmodium Maria E
Francia et al. Cilia (2016) 5:3 DOI 10.1186/s13630-016-0025-5 Cilia REVIEW Open Access Basal body structure and composition in the apicomplexans Toxoplasma and Plasmodium Maria E. Francia1* , Jean‑Francois Dubremetz2 and Naomi S. Morrissette3 Abstract The phylum Apicomplexa encompasses numerous important human and animal disease-causing parasites, includ‑ ing the Plasmodium species, and Toxoplasma gondii, causative agents of malaria and toxoplasmosis, respectively. Apicomplexans proliferate by asexual replication and can also undergo sexual recombination. Most life cycle stages of the parasite lack flagella; these structures only appear on male gametes. Although male gametes (microgametes) assemble a typical 9 2 axoneme, the structure of the templating basal body is poorly defined. Moreover, the rela‑ tionship between asexual+ stage centrioles and microgamete basal bodies remains unclear. While asexual stages of Plasmodium lack defined centriole structures, the asexual stages of Toxoplasma and closely related coccidian api‑ complexans contain centrioles that consist of nine singlet microtubules and a central tubule. There are relatively few ultra-structural images of Toxoplasma microgametes, which only develop in cat intestinal epithelium. Only a subset of these include sections through the basal body: to date, none have unambiguously captured organization of the basal body structure. Moreover, it is unclear whether this basal body is derived from pre-existing asexual stage centrioles or is synthesized de novo. Basal bodies in Plasmodium microgametes are thought to be synthesized de novo, and their assembly remains ill-defined. Apicomplexan genomes harbor genes encoding δ- and ε-tubulin homologs, potentially enabling these parasites to assemble a typical triplet basal body structure. -

Implications of Habitat Restoration for Bumble Bee Population Dynamics, Foraging Ecology, and Epidemiology
IMPLICATIONS OF HABITAT RESTORATION FOR BUMBLE BEE POPULATION DYNAMICS, FORAGING ECOLOGY, AND EPIDEMIOLOGY By Knute Baldwin Gundersen A DISSERTATION Submitted to Michigan State University in partial fulfillment of the requirements for the degree of Entomology—Doctor of Philosophy Ecology, Evolutionary Biology and Behavior—Dual Major 2018 ABSTRACT IMPLICATIONS OF HABITAT RESTORATION FOR BUMBLE BEE POPULATION DYNAMICS, FORAGING ECOLOGY, AND EPIDEMIOLOGY By Knute Baldwin Gundersen Many insects provide valuable ecosystem services, including those that support our food supply. Beneficial insects such as pollinators fulfill part of this role by contributing to approximately one third of the global food crop production. Over the past few decades, pollinators have faced declining populations due to a variety of factors such as agricultural intensification, lack of floral and nesting resources, and disease. One method used in agricultural settings to help sustain pollinator populations is designating unfarmed habitat such as ditches and field margins for habitat enhancement in the form of hedgerows and wildflower strips. These floristically rich areas can be tailored to bloom both before and after crop bloom to help sustain pollinators during the time when crops are not in bloom. In turn, bee populations can benefit from the consistent availability of resources in these areas of habitat enhancement. This dissertation explores how habitat enhancement affects nesting density of a common wild pollinator, Bombus impatiens . Further, this research also aims to determine how foraging preferences change and how bumble bee disease transmission and prevalence respond to habitat enhancement. Research was conducted at 15 commercial highbush blueberry ( Vaccinium corymbosum ) fields in southwest Michigan containing either no restoration, a newly planted restoration, or a mature (5-8 year old) restoration in the field margin from 2015 to 2017. -

New Species of Choleoeimeria (Apicomplexa: Eimeriidae) from The
FOLIA PARASITOLOGICA 53: 91–97, 2006 New species of Choleoeimeria (Apicomplexa: Eimeriidae) from the veiled chameleon, Chamaeleo calyptratus (Sauria: Chamaeleonidae), with taxonomic revision of eimerian coccidia from chameleons Michal Sloboda1 and David Modrý1,2 1Department of Parasitology, University of Veterinary and Pharmaceutical Sciences Brno, Palackého 1–3, 612 42 Brno, Czech Republic; 2Institute of Parasitology, Academy of Sciences of the Czech Republic, Branišovská 31, 370 05 České Budějovice, Czech Republic Key words: Coccidia, Eimeriorina, Choleoeimeria, taxonomy Abstract. Coprological examination of 71 samples from a breeding colony of veiled chameleons, Chamaeleo calyptratus Duméril et Duméril, 1851, revealed a presence of two species of coccidia. In 100% of the samples examined, oocysts of Isospora jaracimrmani Modrý et Koudela, 1995 were detected. A new coccidian species, Choleoeimeria hirbayah sp. n., was discovered in 32.4% of samples from the colony. Its oocysts are tetrasporocystic, cylindrical, 28.3 (25–30) × 14.8 (13.5–17.5) µm, with smooth, bilayered, ~1 µm thick wall. Sporocysts are dizoic, ovoidal to ellipsoidal, 10.1 (9–11) × 6.9 (6–7.5) µm, sporocyst wall is composed of two plates joined by a meridional suture. Endogenous development is confined to the epithelium of the gall blad- der, with infected cells being typically displaced from the epithelium layer towards lumen. A taxonomic revision of tetrasporo- cystic coccidia in the Chamaeleonidae is provided. In reptilian hosts, monoxenous coccidia, namely Ei- of individuals are kept in North America and Europe meria Schneider, 1875 sensu lato and Isospora Schnei- (Nečas 2004). der, 1881 represent commonly diagnosed protozoans of Until now, Isospora jaracimrmani Modrý et Kou- remarkable diversity (Barnard and Upton 1994, Greiner dela, 1995 is the only eimerian coccidium described 2003). -

Supplementary Table S2: New Taxonomic Assignment of Sequences of Basal Fungal Lineages
Supplementary Table S2: New taxonomic assignment of sequences of basal fungal lineages. Fungal sequences were subjected to BLAST-N analysis and checked for their taxonomic placement in the eukaryotic guide-tree of the SILVA release 111. Sequences were classified depending on combined results from the methods mentioned above as well as literature searches. Accession Name New classification Clustering of the sequence in the Best BLAST-N hit number based on combined results eukaryotic guide tree of SILVA Name Accession number E.value Identity AB191431 Uncultured fungus Chytridiomycota Chytridiomycota Basidiobolus haptosporus AF113413.1 0.0 91 AB191432 Unculltured eukaryote Blastocladiomycota Blastocladiomycota Rhizophlyctis rosea NG_017175.1 0.0 91 AB252775 Uncultured eukaryote Chytridiomycota Chytridiomycota Blastocladiales sp. EF565163.1 0.0 91 AB252776 Uncultured eukaryote Fungi Nucletmycea_Fonticula Rhizophydium sp. AF164270.2 0.0 87 AB252777 Uncultured eukaryote Chytridiomycota Chytridiomycota Basidiobolus haptosporus AF113413.1 0.0 91 AB275063 Uncultured fungus Chytridiomycota Chytridiomycota Catenomyces sp. AY635830.1 0.0 90 AB275064 Uncultured fungus Chytridiomycota Chytridiomycota Endogone lactiflua DQ536471.1 0.0 91 AB433328 Nuclearia thermophila Nuclearia Nucletmycea_Nuclearia Nuclearia thermophila AB433328.1 0.0 100 AB468592 Uncultured fungus Basal clone group I Chytridiomycota Physoderma dulichii DQ536472.1 0.0 90 AB468593 Uncultured fungus Basal clone group I Chytridiomycota Physoderma dulichii DQ536472.1 0.0 91 AB468594 Uncultured -

Identification of a Novel Fused Gene Family Implicates Convergent
Chen et al. BMC Genomics (2018) 19:306 https://doi.org/10.1186/s12864-018-4685-y RESEARCH ARTICLE Open Access Identification of a novel fused gene family implicates convergent evolution in eukaryotic calcium signaling Fei Chen1,2,3, Liangsheng Zhang1, Zhenguo Lin4 and Zong-Ming Max Cheng2,3* Abstract Background: Both calcium signals and protein phosphorylation responses are universal signals in eukaryotic cell signaling. Currently three pathways have been characterized in different eukaryotes converting the Ca2+ signals to the protein phosphorylation responses. All these pathways have based mostly on studies in plants and animals. Results: Based on the exploration of genomes and transcriptomes from all the six eukaryotic supergroups, we report here in Metakinetoplastina protists a novel gene family. This family, with a proposed name SCAMK,comprisesSnRK3 fused calmodulin-like III kinase genes and was likely evolved through the insertion of a calmodulin-like3 gene into an SnRK3 gene by unequal crossover of homologous chromosomes in meiosis cell. Its origin dated back to the time intersection at least 450 million-year-ago when Excavata parasites, Vertebrata hosts, and Insecta vectors evolved. We also analyzed SCAMK’s unique expression pattern and structure, and proposed it as one of the leading calcium signal conversion pathways in Excavata parasite. These characters made SCAMK gene as a potential drug target for treating human African trypanosomiasis. Conclusions: This report identified a novel gene fusion and dated its precise fusion time -

The Planktonic Protist Interactome: Where Do We Stand After a Century of Research?
bioRxiv preprint doi: https://doi.org/10.1101/587352; this version posted May 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Bjorbækmo et al., 23.03.2019 – preprint copy - BioRxiv The planktonic protist interactome: where do we stand after a century of research? Marit F. Markussen Bjorbækmo1*, Andreas Evenstad1* and Line Lieblein Røsæg1*, Anders K. Krabberød1**, and Ramiro Logares2,1** 1 University of Oslo, Department of Biosciences, Section for Genetics and Evolutionary Biology (Evogene), Blindernv. 31, N- 0316 Oslo, Norway 2 Institut de Ciències del Mar (CSIC), Passeig Marítim de la Barceloneta, 37-49, ES-08003, Barcelona, Catalonia, Spain * The three authors contributed equally ** Corresponding authors: Ramiro Logares: Institute of Marine Sciences (ICM-CSIC), Passeig Marítim de la Barceloneta 37-49, 08003, Barcelona, Catalonia, Spain. Phone: 34-93-2309500; Fax: 34-93-2309555. [email protected] Anders K. Krabberød: University of Oslo, Department of Biosciences, Section for Genetics and Evolutionary Biology (Evogene), Blindernv. 31, N-0316 Oslo, Norway. Phone +47 22845986, Fax: +47 22854726. [email protected] Abstract Microbial interactions are crucial for Earth ecosystem function, yet our knowledge about them is limited and has so far mainly existed as scattered records. Here, we have surveyed the literature involving planktonic protist interactions and gathered the information in a manually curated Protist Interaction DAtabase (PIDA). In total, we have registered ~2,500 ecological interactions from ~500 publications, spanning the last 150 years. -

Prevalence of Cryptosporidium Spp. \(Eucoccidiorida
Article available at http://www.parasite-journal.org or http://dx.doi.org/10.1051/parasite/2007144335 PREVALENCE OF CRYPTOSPORIDIUM SPP. (EUCOCCIDIORIDA: CRYPTOSPORIIDAE) IN SEVEN SPECIES OF FARM ANIMALS IN TUNISIA SOLTANE R.*, GUYOT K.**, DEI-CAS E.** & AYADI A.* Summary: Résumé : PRÉVALENCE DE CRYPTOSPORIDIUM SPP. (EUCOCCIDIORIDA : CRYPTOSPORIIDAE) CHEZ SEPT ESPÈCES D’ANIMAUX DE FERME EN TUNISIE 1,001 faecal samples were obtained from 89 sheep (lambs and adult), 184 goats, 190 horses, 178 rabbits, 110 camels, 1001 prélèvements fécaux ont été obtenus à partir de 89 moutons, 200 broiler chicken and 50 turkeys housed in farms from different 184 chèvres, 190 chevaux, 178 lapins, 110 chameaux, localities in Tunisia. All samples were analysed for 200 poulets et 50 dindes élevés dans des fermes de différentes Cryptosporidium oocysts by microscopic examination of smears localités en Tunisie. Tous les prélèvements ont été analysés pour la stained by modified Ziehl Neelsen technique. The parasite was recherche de Cryptosporidium par examen microscopique des detected in ten lambs and adult sheep (11.2 %) and nine broiler frottis colorés au Ziehl Neelsen modifié. Le parasite a été détecté chicken (4.5 %). Molecular characterization, performed in four chez dix ovins (11,2 %) et neuf poulets (4,5 %). La caractérisation animals, identified C. bovis in three lambs and C. meleagridis in moléculaire réalisée pour quatre isolats a identifié C. bovis chez one broiler chicken. This work is the first report on trois agneaux et C. meleagridis chez un poulet. Ce travail est le Cryptosporidium in farm animals in Tunisia. premier rapport sur Cryptosporidium chez des animaux de ferme en Tunisie. -

Journal of Parasitology
Journal of Parasitology Eimeria taggarti n. sp., a Novel Coccidian (Apicomplexa: Eimeriorina) in the Prostate of an Antechinus flavipes --Manuscript Draft-- Manuscript Number: 17-111R1 Full Title: Eimeria taggarti n. sp., a Novel Coccidian (Apicomplexa: Eimeriorina) in the Prostate of an Antechinus flavipes Short Title: Eimeria taggarti n. sp. in Prostate of Antechinus flavipes Article Type: Regular Article Corresponding Author: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc University of Melbourne Melbourne, Victoria AUSTRALIA Corresponding Author Secondary Information: Corresponding Author's Institution: University of Melbourne Corresponding Author's Secondary Institution: First Author: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc First Author Secondary Information: Order of Authors: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc Joanne Maree Devlin, BVSc(Hons), MVPHMgt, PhD Liliana Tatarczuch David Augustine Taggart David J Schultz Jenny A Charles Ian Beveridge Order of Authors Secondary Information: Abstract: A novel coccidian species was discovered in the prostate of an Antechinus flavipes (yellow-footed antechinus) in South Australia, during the period of post-mating male antechinus immunosuppression and mortality. This novel coccidian is unusual because it develops extra-intestinally and sporulates endogenously within the prostate gland of its mammalian host. Histological examination of prostatic tissue revealed dense aggregations of spherical and thin-walled tetrasporocystic, dizoic sporulated coccidian oocysts within tubular lumina, with unsporulated oocysts and gamogonic stages within the cytoplasm of glandular epithelial cells. This coccidian was observed occurring concurrently with dasyurid herpesvirus 1 infection of the antechinus' prostate. Eimeria- specific 18S small subunit ribosomal DNA PCR amplification was used to obtain a partial 18S rDNA nucleotide sequence from the antechinus coccidian. -

ABSTRACT Gregarine Parasitism in Dragonfly Populations of Central
ABSTRACT Gregarine Parasitism in Dragonfly Populations of Central Texas with an Assessment of Fitness Costs in Erythemis simplicicollis Jason L. Locklin, Ph.D. Mentor: Darrell S. Vodopich, Ph.D. Dragonfly parasites are widespread and frequently include gregarines (Phylum Apicomplexa) in the gut of the host. Gregarines are ubiquitous protozoan parasites that infect arthropods worldwide. More than 1,600 gregarine species have been described, but only a small percentage of invertebrates have been surveyed for these apicomplexan parasites. Some consider gregarines rather harmless, but recent studies suggest otherwise. Odonate-gregarine studies have more commonly involved damselflies, and some have considered gregarines to rarely infect dragonflies. In this study, dragonfly populations were surveyed for gregarines and an assessment of fitness costs was made in a common and widespread host species, Erythemis simplicicollis. Adult dragonfly populations were surveyed weekly at two reservoirs in close proximity to one another and at a flow-through wetland system. Gregarine prevalences and intensities were compared within host populations between genders, among locations, among wing loads, and through time. Host fitness parameters measured included wing load, egg size, clutch size, and total egg count. Of the 37 dragonfly species surveyed, 14 species (38%) hosted gregarines. Thirteen of those species were previously unreported as hosts. Gregarine prevalences ranged from 2% – 52%. Intensities ranged from 1 – 201. Parasites were aggregated among their hosts. Gregarines were found only in individuals exceeding a minimum wing load, indicating that gregarines are likely not transferred from the naiad to adult during emergence. Prevalence and intensity exhibited strong seasonality during both years at one of the reservoirs, but no seasonal trend was detected at the wetland. -

An Intestinal Gregarine of Nothria Conchylega (Polychaeta, Onuphidae)
Journal of Invertebrate Pathology 104 (2010) 172–179 Contents lists available at ScienceDirect Journal of Invertebrate Pathology journal homepage: www.elsevier.com/locate/jip Description of Trichotokara nothriae n. gen. et sp. (Apicomplexa, Lecudinidae) – An intestinal gregarine of Nothria conchylega (Polychaeta, Onuphidae) Sonja Rueckert *, Brian S. Leander Canadian Institute for Advanced Research, Program in Integrated Microbial Biodiversity, Departments of Botany and Zoology, University of British Columbia, #3529 – 6270 University Blvd., Vancouver, BC, Canada V6T 1Z4 article info abstract Article history: The trophozoites of a novel gregarine apicomplexan, Trichotokara nothriae n. gen. et sp., were isolated and Received 12 November 2009 characterized from the intestines of the onuphid tubeworm Nothria conchylega (Polychaeta), collected at Accepted 11 March 2010 20 m depth from the North-eastern Pacific Coast. The trophozoites were 50–155 lm long with a mid-cell Available online 23 March 2010 indentation that formed two prominent bulges (anterior bulge, 14–48 lm wide; posterior bulge, 15– 55 lm wide). Scanning electron microscopy (SEM) demonstrated that approximately 400 densely packed, Keywords: longitudinal epicytic folds (5 folds/lm) inscribe the surface of the trophozoites, and a prominently elon- Alveolata gated mucron (14–60 lm long and 6–12 lm wide) was covered with hair-like projections (mean length, Apicomplexa 1.97 m; mean width, 0.2 m at the base). Although a septum occurred at the junction between the cell Lecudinidae l l Lecudina proper and the mucron in most trophozoites, light microscopy (LM) demonstrated that the cell proper Parasite extended into the core of the mucron as a thin prolongation. -
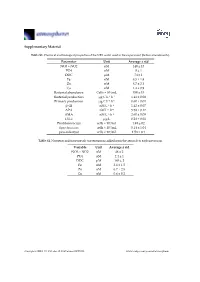
Supplementary Material Parameter Unit Average ± Std NO3 + NO2 Nm
Supplementary Material Table S1. Chemical and biological properties of the NRS water used in the experiment (before amendments). Parameter Unit Average ± std NO3 + NO2 nM 140 ± 13 PO4 nM 8 ± 1 DOC μM 74 ± 1 Fe nM 8.5 ± 1.8 Zn nM 8.7 ± 2.1 Cu nM 1.4 ± 0.9 Bacterial abundance Cells × 104/mL 350 ± 15 Bacterial production μg C L−1 h−1 1.41 ± 0.08 Primary production μg C L−1 h−1 0.60 ± 0.01 β-Gl nM L−1 h−1 1.42 ± 0.07 APA nM L−1 h−1 5.58 ± 0.17 AMA nM L−1·h−1 2.60 ± 0.09 Chl-a μg/L 0.28 ± 0.01 Prochlorococcus cells × 104/mL 1.49 ± 02 Synechococcus cells × 104/mL 5.14 ± 1.04 pico-eukaryot cells × 103/mL 1.58 × 0.1 Table S2. Nutrients and trace metals concentrations added from the aerosols to each mesocosm. Variable Unit Average ± std NO3 + NO2 nM 48 ± 2 PO4 nM 2.4 ± 1 DOC μM 165 ± 2 Fe nM 2.6 ± 1.5 Zn nM 6.7 ± 2.5 Cu nM 0.6 ± 0.2 Atmosphere 2019, 10, 358; doi:10.3390/atmos10070358 www.mdpi.com/journal/atmosphere Atmosphere 2019, 10, 358 2 of 6 Table S3. ANOVA test results between control, ‘UV-treated’ and ‘live-dust’ treatments at 20 h or 44 h, with significantly different values shown in bold. ANOVA df Sum Sq Mean Sq F Value p-value Chl-a 20 H 2, 6 0.03, 0.02 0.02, 0 4.52 0.0634 44 H 2, 6 0.02, 0 0.01, 0 23.13 0.002 Synechococcus Abundance 20 H 2, 7 8.23 × 107, 4.11 × 107 4.11 × 107, 4.51 × 107 0.91 0.4509 44 H 2, 7 5.31 × 108, 6.97 × 107 2.65 × 108, 1.16 × 107 22.84 0.0016 Prochlorococcus Abundance 20 H 2, 8 4.22 × 107, 2.11 × 107 2.11 × 107, 2.71 × 106 7.77 0.0216 44 H 2, 8 9.02 × 107, 1.47 × 107 4.51 × 107, 2.45 × 106 18.38 0.0028 Pico-eukaryote -

Evidence for and Against Deformed Wing Virus Spillover from Honey Bees to Bumble Bees: a Reverse Genetic Analysis Olesya N
www.nature.com/scientificreports OPEN Evidence for and against deformed wing virus spillover from honey bees to bumble bees: a reverse genetic analysis Olesya N. Gusachenko1*, Luke Woodford1, Katharin Balbirnie‑Cumming1, Eugene V. Ryabov2 & David J. Evans1* Deformed wing virus (DWV) is a persistent pathogen of European honey bees and the major contributor to overwintering colony losses. The prevalence of DWV in honey bees has led to signifcant concerns about spillover of the virus to other pollinating species. Bumble bees are both a major group of wild and commercially‑reared pollinators. Several studies have reported pathogen spillover of DWV from honey bees to bumble bees, but evidence of a sustained viral infection characterized by virus replication and accumulation has yet to be demonstrated. Here we investigate the infectivity and transmission of DWV in bumble bees using the buf-tailed bumble bee Bombus terrestris as a model. We apply a reverse genetics approach combined with controlled laboratory conditions to detect and monitor DWV infection. A novel reverse genetics system for three representative DWV variants, including the two master variants of DWV—type A and B—was used. Our results directly confrm DWV replication in bumble bees but also demonstrate striking resistance to infection by certain transmission routes. Bumble bees may support DWV replication but it is not clear how infection could occur under natural environmental conditions. Deformed wing virus (DWV) is a widely established pathogen of the European honey bee, Apis mellifera. In synergistic action with its vector—the parasitic mite Varroa destructor—it has had a devastating impact on the health of honey bee colonies globally1,2.