Chem 355 Lab Manual Fall, 2005 Minnesota State University Moorhead Department of Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

LAB: One Tube Reaction Part 1
AP Chemistry LAB: One Tube Reaction Part 1 Objective: To monitor and document the chemical changes occurring in a single test tube containing a predetermined mixture of chemicals. Materials: test tube, iron nail, sodium chloride, copper (II) sulfate, distilled water, glass stirring rod, tissue paper, parafilm wax Procedures: 1. Label a test tube to identify your group 2. Fill approx. 1/3 of the test tube with copper (II) sulfate crystals. Gently tap the tube to allow the crystals to settle. 3. Using a glass stirring rod, carefully cover the crystals with a layer of Kimwipe tissue paper. 4. Slowly, and with as little disturbance as possible, add enough distilled water to just cover the paper and blue crystals. 5. Repeat the process for the sodium chloride, filling approx. 1/3 of the test tube. Gently tap the tube to allow the crystals to settle. 6. Push more tissue paper into the test tube on top of the white crystals. 7. Add enough water to cover the tissue paper and white crystals. 8. Obtain an iron nail and expose the surface by rubbing with sand paper. 9. Carefully slide the nail into the test tube. 10. Continue adding water until the nail is completely covered. 11. Cover the test tube with parafilm wax and record your Day 1 observations. 12. Return the test tube to the test tube rack. 13. Continue to monitor and record your observations for the next several days, as indicated in the data table. Data: 1. Complete the NFPA label for the two chemicals used in this lab. -

Equipment Detailsr07
Lab Equipment Details Lab Equipment Glass Flasks 150ml 250ml 500ml Lab Equipment Glass Beakers 150ml 250ml 500ml Lab Equipment Glassware But once removed, only the cap stays highlighted. Droppers critical to Lab An activated Dropper coursework can be found highlights the entire bottle. already in the workspace. Dropper Dropper Dropper Activated In Use Lab Equipment Gastight Syringe Small A pre-filled gastight syringe can be used with a NMR tube to safely fill through the top in preparation for use with the NMR spectrometer. Gastight Syringe NMR Tube with Holder with Holder Lab Equipment NMR Tube Spinner An NMR Tube filled with gas for use with the NMR Spectrometer Simply use the NMR Tube with needs to be inserted into the the Tube Spinner. Spinner before it can be used. NMR Tube NMR Tube Spinner NMR Tube Spinner with Holder with NMR Tube inserted Once the Tube slotted into the holder is inserted into the top part of the spectrometer, users can type in a number for Lab Equipment the frequency they’d like to scan. NMR Spectrometer XL The NMR Tube holder can then be slotted into the highlighted tube on the NMR Spectrometer. NMR Tube filled with appropriate substance is slotted (used with) in the holder. Lift will perpare the Spectrometer for the tube holder insertion. Scan No. Allows the user to change the frequency at which the tube is scanned The NMR Tube holder can then be slotted into the top tube on Lab Equipment the NMR Spectrometer. NMR Spectrometer XL NMR Tube filled with appropriate substance is slotted (used with) in the holder. -

Documm Mgson!
Documm mgson! ED 174 412 SE 027 981 TITLE Biomedical Science, Unit I: Respiration inHealth and Medicine. Respiratory Anatomy, Physiology and Pathology; The Behavior of Gases;Introductory Chemistry; and Air Pollution. Laboratory Manual. Revised Version, 1975. INSTITUTION Biomedical Interdisciplinary Curriculum Project, Berkeley, Calif. SPONS AGENCY National Science Foundation, Washington, D.C. PUB DATE 75 NOTE 181p.; For related documents, see SE 027978-999 and SE 028 510-516; Not available in hard copydue to copyright restrictions EDRS PRICE MF01 Plus Postage. PC Not Available from EDRS. DESCRIpTCRS Chemistry; Environment; Health; *HealthEducation; Higher Education; *Laboratory ExFeriments; *Laboratory Techniques; *Laboratory Training;Medical Education; Pathology; Physics; *Physiology;*Science Education; Secondary Education IDENTIFIERS *Respiration ABSTItACT Designed to accompany the student text on respiration, this manual presents instructions on the use of labcra tcry equipment and presents various experimentsdealing with the concepts presented in the text. Thirty=nine laboratoryactivities ' are de scribed. Laboratory activities are dividedinto several parts, each Part covering a specific experiment dealing with the concept covered by the activity. Each experimentincludes a description of materials, procedures, and discurision questions. (RE) ********************************************************************** Reproductions supplied by EDRS are the bestthat can be made from the original document. 4********************M414141#***********4141*****4141***********41#********4 -
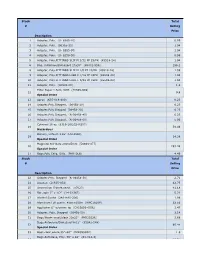
Stock Total # Selling Price 1 Adapter, Poly. (U- 6365-10)
Stock Total # Selling Price Description 1 Adapter, Poly. (U- 6365-10) 0.99 2 Adapter, Poly. (06365-22) 1.04 3 Adapter, Poly. (U- 6365-30) 1.04 4 Adapter, Poly. (U- 6359-50) 0.99 5 Adapter, Poly,FITTINGS SLIP M 3/32 PP 25/PK (45518-24) 1.04 6 Bag, Autoclave/Biohazard 24x30'' (95042-556) 156.1 8 Adapter, Poly.FITTINGS SLIP M 1/8 PP 25/PK (45518-26) 1.59 9 Adapter, Poly.FITTINGS LUER F 1/16 PP 25PK (45508-00) 1.04 10 Adapter, Poly.FITTINGS LUER F 3/32 PP 25PK (45508-02) 1.04 11 Adapter, Poly. (30800-08) 1.3 Filter Paper 1.5cm, VWR (28309-989) 12 9.9 Special Order 13 Apron (635-018-400) 0.23 14 Adapter,Poly.Stepped, (06458-10) 6.23 15 Adapter,Poly.Stepped (06458-20) 6.23 16 Adapter,Poly.Stepped, (N-06458-40) 6.23 17 Adapter,Poly.Stepped, (N-06458-60) 5.06 Cytoseal 16 oz (8310-16/23244257) 18 58.49 Hazardous Battery, Lithium 3.6V (LS14500) 19 14.38 Special Order Magnetic Stir Bars 2mmx5mm (58948-377) 20 293.48 Special Order 21 Bags,Poly Cello,10 lb. (PKR10Lb) 4.49 Stock Total # Selling Price Description 22 Adapter,Poly. Stepped (N-06458-30) 2.71 24 Alconox (21835-032) 42.75 25 Ammonium Sulfate,certif. (A7023) 41.18 26 Bar, spin 2'' x 1/2'' (14-51367) 5.22 27 Alcohol Swabs (248-HAS-200) 1.96 28 Aluminum Foil comm. 45cmx100m (WPC1810P) 32.56 29 Applicator 6'' w/cotton tip (CA10806-000L) 1.47 30 Adapter, Poly. -

Laboratory Equipment AP
\ \\ , f ?7-\ Watch glass 1 Crucible and cover Evaporating dish Pneumatlo trough Beaker Safety goggles Florence Wide-mouth0 Plastic wash Dropper Funnel flask collecting bottle pipet Edenmeyer Rubber stoppers bottle flask € ....... ">. ÿ ,, Glass rod with niohrome wire Scoopula (for flame re,sting) CruoiNe tongs Rubber ubing '1 ,v .... Test-tube brush square Wire gau ÿ "\ file Burner " Tripod Florence flask: glass; common sizes are 125 mL, 250 mL, 500 .d Beaker: glass or plastic; common sizes are 50 mL, mL; maybe heated; used in making and for storing solutions. 100 mL, 250 mL, 400 mL; glass beakers maybe heated. oÿ Buret: glass; common sizes are 25 mL and 50 mL; used to Forceps: metal; used to hold or pick up small objects. Funnel: glass or plastic; common size holds 12.5-cm diameter measure volumes of solutions in titrafions. Ceramic square: used under hot apparatus or glassware. filter paper. Gas burner: constructed of metal; connected to a gas supply Clamps" the following types of clamps may be fastened to with rubber tubing; used to heat chemicals (dry or in solution) support apparatus: buret/test-tube clamp, clamp holder, double buret clamp, ring clamp, 3-pronged jaw clamp. in beakers, test tubes, and crucibles. Gas collecting tube: glass; marked in mL intervals; used to 3: Clay triangle: wire frame with porcelain supports; used to o} support a crucible. measure gas volumes. Glass rod with nichrome wire: used in flame tests. Condenser: glass; used in distillation procedures. Q. Crucible and cover: porcelain; used to heat small amounts of Graduated cylinder: glass or plastic; common sizes are 10 mL, 50 mL, 100 mL; used to measure approximate volumes; must solid substances at high temperatures. -

Lab # 4: Separation of a Mixture
Name: Date Completed: Lab Partner(s): Lab # 4: Separation of a Mixture Lab Accelerated Chemistry 1 Objective You will be given a mixture containing sodium chloride (NaCl, table salt), benzoic acid (C7H6O2, a common food preservative), and silicon dioxide (SiO2, sand). Your goal is to separate the substances and determine the percent of each in the original mixture. Here is the separation scheme: Mixture (NaCl, benzoic acid, sand) Extract with 100°C H O 2 Decant residue supernatant Sand (wet) NaCl, benzoic acid Cool Filter evaporate H O 2 residue filtrate Sand (dry) Benzoic acid (wet) NaCl (solution) evaporate H2O Benzoic acid (dry) Introduction Mixtures are not unique to chemistry; we use and consume them on a daily basis. The beverages we drink each morning, the fuel we use in our automobiles, and the ground we walk on are mixtures. Very few materials we encounter are pure. Any material made up of two or more substances that are not chemically combined is a mixture. The isolation of pure components of a mixture requires the separation of one component from another. Chemists have developed techniques for doing this. These methods take advantage of the differences in physical properties of the components. The techniques to be demonstrated in this laboratory are the following: Extraction. This uses a solvent to selectively dissolve one component of the solid mixture. With this technique, a soluble solid can be separated from an insoluble solid. Decantation. This separates a liquid from insoluble solid sediment by carefully pouring the liquid from the solid without disturbing the solid. Recrystallization. -

Used for Moving Beakers Off of Hot Surfaces
Lab Equipment and Use Review Sheet Glassware Function Glassware Function Glassware Function Running Accurately Running reactions, measuring/ reactions, heating chemicals mixing mixing delivering chemicals – chemicals, volumes of easier for mixing Beaker heating Buret liquids than beakers chemicals Erlenmeyer Flask Used in Running Used to mix vacuum reactions, chemicals to heating filtration chemicals accurately mixing determine chemicals – concentration; Volumetric easier for contains exact Florence Flask Flask Filter Flask mixing than volumes beakers Used for Used for Used for mixing filtering and accurately and heating chemicals and for adding measuring running chemicals the volume reactions– without of liquids. smaller quantities Funnel Graduated spilling Test Tube than beakers and Cylinder flasks. Holding For stirring For storing Chemicals, Glass Stirring Rod chemicals small amounts covering of chemicals Watch Glass beakers during Sample Vial heating For adding Used to small evaporate amounts of Used for Evaporating liquids chemicals – lighting burner Dish usually by Plastic Pipets drops Used to add Utility Clamp – deionized used to hold water; to add objects on a solvents for ring stand. cleaning of Iron ring – Squirt Bottle beakers and used to hold other objects above glassware. a Bunsen burner flame. Ring Stand Ring stand – Beaker Tongs with Utility used to hold Clamp and various Iron Ring objects. Equipment Function Equipment Function Equipment Function Heat source in Used to grind Used to clean the chemistry chemicals into glassware lab. Uses powder natural gas. Mortar and Pestle Test tube brush/beaker Bunsen Burner brush Used to hold a Used to Used to crucible above transport hot strongly heat a flame – used Crucible tongs crucibles and substances Clay triangle in conjunction to remove Crucible and above a flame with iron ring their covers Cover Used to hold Used to handle a Used in group of test single test tube. -

1455189355674.Pdf
THE STORYTeller’S THESAURUS FANTASY, HISTORY, AND HORROR JAMES M. WARD AND ANNE K. BROWN Cover by: Peter Bradley LEGAL PAGE: Every effort has been made not to make use of proprietary or copyrighted materi- al. Any mention of actual commercial products in this book does not constitute an endorsement. www.trolllord.com www.chenaultandgraypublishing.com Email:[email protected] Printed in U.S.A © 2013 Chenault & Gray Publishing, LLC. All Rights Reserved. Storyteller’s Thesaurus Trademark of Cheanult & Gray Publishing. All Rights Reserved. Chenault & Gray Publishing, Troll Lord Games logos are Trademark of Chenault & Gray Publishing. All Rights Reserved. TABLE OF CONTENTS THE STORYTeller’S THESAURUS 1 FANTASY, HISTORY, AND HORROR 1 JAMES M. WARD AND ANNE K. BROWN 1 INTRODUCTION 8 WHAT MAKES THIS BOOK DIFFERENT 8 THE STORYTeller’s RESPONSIBILITY: RESEARCH 9 WHAT THIS BOOK DOES NOT CONTAIN 9 A WHISPER OF ENCOURAGEMENT 10 CHAPTER 1: CHARACTER BUILDING 11 GENDER 11 AGE 11 PHYSICAL AttRIBUTES 11 SIZE AND BODY TYPE 11 FACIAL FEATURES 12 HAIR 13 SPECIES 13 PERSONALITY 14 PHOBIAS 15 OCCUPATIONS 17 ADVENTURERS 17 CIVILIANS 18 ORGANIZATIONS 21 CHAPTER 2: CLOTHING 22 STYLES OF DRESS 22 CLOTHING PIECES 22 CLOTHING CONSTRUCTION 24 CHAPTER 3: ARCHITECTURE AND PROPERTY 25 ARCHITECTURAL STYLES AND ELEMENTS 25 BUILDING MATERIALS 26 PROPERTY TYPES 26 SPECIALTY ANATOMY 29 CHAPTER 4: FURNISHINGS 30 CHAPTER 5: EQUIPMENT AND TOOLS 31 ADVENTurer’S GEAR 31 GENERAL EQUIPMENT AND TOOLS 31 2 THE STORYTeller’s Thesaurus KITCHEN EQUIPMENT 35 LINENS 36 MUSICAL INSTRUMENTS -

GENERAL CHEMISTRY 101 LABORATORY MANUAL an Inquiry Approach Through an Environmental Awareness
East Los Angeles College Department of Chemistry GENERAL CHEMISTRY 101 LABORATORY MANUAL An Inquiry Approach through an Environmental Awareness The following laboratories have been compiled and adapted by Alan Khuu, M.S. & Armando Rivera, Ph.D. 1 Table of Contents I. Chemical Safety in the Laboratory ............................................................. 4 II. Green Chemistry Twelve (12) Principles of EPA Green Chemistry ........................................ 7 III. Unit 1 Water ............................................................................................................. 9 IV. Experiment 2 Water Analysis ............................................................................................ 18 V. Experiment 3 Methods of Evaporation and Filtration Used to Separate Mixtures ..................................................................................................... 29 VI. Unit 2 Waste and Recycling.................................................................................... 34 VII. Experiment 4 Waste and Recycling of Aluminum ............................................................ 39 VIII. Experiment 5 Identification of an Unknown Compound Through Mass Correlations ................................................................................................ 46 IX. Experiment 6 Classifying Chemical Reactions Through Copper Reactions ................... 50 X. Experiment 7 Using Titration to Verify the Chemical Formula of Calcium Hydroxide .................................................................................................. -

Chemistry Lab Safety Quiz Example
Chemistry Lab Safety Quiz Example 1. You are excused from wearing goggles in lab: A) if you have permission from your doctor. B) if they are uncomfortable to wear C) if you are finished with the experiment and ready to clean up D) if they offend your sense of style E) none of the above 2. If you do not understand the experimental procedure A) ask your classmate. B) read the experiment many times. C) leave the lab. D) ask the instructor. E) none of the above 3. You are permitted to enter the lab if: A) the door is open and nobody is present. B) the instructor is present. C) the door is open and a group of students are present. D) if the janitor is present with his safety trained dog . E) the door is unlocked 4. Store your books and bags: A) on your lab bench. B) in the aisle between lab benches. C) in the cabinet in the lab. D) under your bench. E) on the top of the instructor’s desk. 1 5. To avoid electric shock when handling electric plugs and equipment: A) your hands must be dry. B) your clothes must cover your torso. C) your food should be kept at a distance. D) your shoes must cover your toes. E) none of the above. 6. Proper footwear in lab is: A) sandals that allow proper ventilation to the feet. B) a comfortable pair of slippers. C) closed shoes that don’t expose any part of the feet D) shoes with low heel. -

Chemistry 2B Lab Manual Standard Operating Procedures Winter Quarter 2018
Chemistry 2B Lab Manual Standard Operating Procedures Winter Quarter 2018 Department of Chemistry University of California - Davis Davis, CA 95616 Student Name Locker # Laboratory Information Teaching Assistant’s Name Laboratory Section Number Laboratory Room Number Dispensary Room Number 1060 Sciences Lab Building Location of Safety Equipment Nearest to Your Laboratory Safety Shower Eye Wash Fountain Fire Extinguisher Fire Alarm Safety Chemicals Revision Date 12/1/2017 Preface Chemistry is an experimental science. Thus, it is important that students of chemistry do experiments in the laboratory to more fully understand that the theories they study in lecture and in their textbook are developed from the critical evaluation of experimental data. The laboratory can also aid the student in the study of the science by clearly illustrating the principles and concepts involved. Finally, laboratory experimentation allows students the opportunity to develop techniques and other manipulative skills that students of science must master. The faculty of the Chemistry Department at UC Davis clearly understands the importance of laboratory work in the study of chemistry. The Department is committed to this component of your education and hopes that you will take full advantage of this opportunity to explore the science of chemistry. A unique aspect of this laboratory program is that a concerted effort has been made to use environmentally less toxic or non-toxic materials in these experiments. This was not only done to protect students but also to lessen the impact of this program upon the environment. This commitment to the environment has presented an enormous challenge, as many traditional experiments could not be used due to the negative impact of the chemicals involved. -

Laboratory Glassware
This week we are launching Wikivoyage . Join us in creating a free travel guide that anyone can edit. Laboratory glassware From Wikipedia, the free encyclopedia Jump to: navigation, search This article needs additional citations for verification. Please help improve this article by adding citations to reliable sources. Unsourced material may be challenged and removed. (February 2011) Three beakers, an Erlenmeyer flask, a graduated cylinder and a volumetric flask Brown glass jars with some clear lab glassware in the background Laboratory glassware refers to a variety of equipment, traditionally made of glass, used for scientific experiments and other work in science, especially in chemistry and biology laboratories. Some of the equipment is now made of plastic for cost, ruggedness, and convenience reasons, but glass is still used for some applications because it is relatively inert, transparent, more heat-resistant than some plastics up to a point, and relatively easy to customize. Borosilicate glasses are often used because they are less subject to thermal stress and are common for reagent bottles. For some applications quartz glass is used for its ability to withstand high temperatures or its transparency in certain parts of the electromagnetic spectrum. In other applications, especially some storage bottles, darkened brown or amber (actinic) glass is used to keep out much of the UV and IR radiation so that the effect of light on the contents is minimized. Special-purpose materials are also used; for example, hydrofluoric acid is stored and used in polyethylene containers because it reacts with glass.[1] For pressurized reaction, heavy-wall glass is used for pressure reactor.