Accurate Taxonomic Assignment of Short Pyrosequencing Reads
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
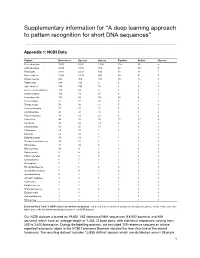
A Deep Learning Approach to Pattern Recognition for Short DNA Sequences”
Supplementary information for “A deep learning approach to pattern recognition for short DNA sequences” Appendix 1: NCBI Data Phylum References Species Genera Families Orders Classes Proteobacteria 7,053 5,061 1,106 158 55 9 Actinobacteria 4,768 3,313 383 68 29 6 Firmicutes 3,814 2,531 499 56 13 7 Bacteroidetes 1,934 1,525 360 39 8 7 Euryarchaeota 834 450 100 28 13 8 Tenericutes 266 195 8 5 4 1 Spirochaetes 146 100 16 6 4 1 Deinococcus-Thermus 118 99 9 3 2 1 Crenarchaeota 113 61 27 8 5 1 Cyanobacteria 113 88 59 30 8 2 Fusobacteria 75 37 10 2 1 1 Thermotogae 70 48 13 5 4 1 Verrucomicrobia 57 53 22 7 4 3 Acidobacteria 45 41 19 5 5 4 Planctomycetes 44 31 23 5 3 2 Chloroflexi 44 35 26 15 12 8 Aquificae 43 32 14 4 2 1 Synergistetes 31 25 15 1 1 1 Chlamydiae 28 18 7 5 2 1 Chlorobi 21 16 5 1 1 1 Deferribacteres 15 11 7 1 1 1 Thermodesulfobacteria 14 12 5 1 1 1 Nitrospirae 11 10 3 1 1 1 Fibrobacteres 10 4 3 3 3 3 Balneolaeota 9 9 4 1 1 1 Chrysiogenetes 6 4 3 1 1 1 Lentisphaerae 6 5 3 3 3 2 Dictyoglomi 5 2 1 1 1 1 Rhodothermaeota 5 5 3 2 1 1 Gemmatimonadetes 5 4 3 2 2 2 Ignavibacteriae 4 2 2 2 1 1 Armatimonadetes 4 3 3 3 3 3 Caldiserica 3 1 1 1 1 1 Calditrichaeota 2 2 1 1 1 1 Thaumarchaeota 2 2 2 2 2 2 Elusimicrobia 2 1 1 1 1 1 Kiritimatiellaeota 1 1 1 1 1 1 Nitrospinae 1 1 1 1 1 1 Extended Data Table 1: NCBI dataset breakdown by phylum. -

7. References
University of Akureyri Department of Natural Resource Science 7. References Ammann, E. C., Reed, L. L., & Durichek, J. J. (1968). Gas consumptions and Growth Rate of Hydrogenomonas eutropha in Continuous Culture. Applied Microbiology , 16, (6), 822-826. Aguiar, P., Beveridge, T. J., & Reysenbach, A.-L. (2004). Sulfurihydrogenibium azorense, sp. nov., a thermophilic hydrogen oxidizing microaerophile from terrestrial hot springs in the Azores. International Journal of Systematic and Evolutionary Microbiology , 54, 33-39. Altschul, S., Gish, W., Miller, W., Myers, E., & Lipman, D. (1990). "Basic local alignment search tool.". J. Mol. Biol. , 215:403-410. Amend, J., & Shock, E. (2001). Energetics of overall metabolic reactions of thermophilic and hyperthermophilic Archaea and Bacteria. FEMS Microbiology Reviews , 25, 175-243. Aragno, M. (1978). Enrichment, isolation and preliminary characterization of a thermophilic, endospore-forming hydrogen bacterium. FEMS Micobiol. Lett. , 3: 13-15. Aragno, M. (1992). The Thermophilic, Aerobic, Hydrogen-Oxidizing (Knallgas) Bacteria. In A. Balows, H. Trüper, M. Dworkin, W. Harder, & K. Schleifer, The Prokaryotes, a handbook on biology of bacteria. 2nd ed. vol. 4 (pp. 3917-3933.). New York: Springer Verlag. Aragno, M., & Schlegel, H. G. (1992). The mesophilic Hydrogen-Oxidizing (Knallgas) Bacteria. In A. Balows, H. Truper, M. Dworkin, W. Harder, & K.-H. Schleifer, The Prokaryotes 2nd. ed. (pp. 344-384). New York: Springer. Ármannson, H. (2002, May 30-31). Erindi á ráðstefnu um málefni veitufyrirtækja . Grænt bókhald í jarðhita- samanburður á útblæstri við aðra orkugjafa . Akureyri, Iceland: Samorka. Bae, S., Kwak, K., Kim, S., Chung, S., & Igarashi, Y. (2001). Isolation and Characterization of CO2-Fixing Hydrogen -Oxidizing Marine 109 University of Akureyri Department of Natural Resource Science Bacteria. -

Characterization of the Uncommon Enzymes from (2004)
OPEN Mannosylglucosylglycerate biosynthesis SUBJECT AREAS: in the deep-branching phylum WATER MICROBIOLOGY MARINE MICROBIOLOGY Planctomycetes: characterization of the HOMEOSTASIS MULTIENZYME COMPLEXES uncommon enzymes from Rhodopirellula Received baltica 13 March 2013 Sofia Cunha1, Ana Filipa d’Avo´1, Ana Mingote2, Pedro Lamosa3, Milton S. da Costa1,4 & Joana Costa1,4 Accepted 23 July 2013 1Center for Neuroscience and Cell Biology, University of Coimbra, 3004-517 Coimbra, Portugal, 2Instituto de Tecnologia Quı´mica Published e Biolo´gica, Universidade Nova de Lisboa, 2780-157 Oeiras, Portugal, 3Centro de Ressonaˆncia Magne´tica Anto´nio Xavier, 7 August 2013 Instituto de Tecnologia Quı´mica e Biolo´gica, Universidade Nova de Lisboa, 2781-901 Oeiras, Portugal, 4Department of Life Sciences, University of Coimbra, Apartado 3046, 3001-401 Coimbra, Portugal. Correspondence and The biosynthetic pathway for the rare compatible solute mannosylglucosylglycerate (MGG) accumulated by requests for materials Rhodopirellula baltica, a marine member of the phylum Planctomycetes, has been elucidated. Like one of the should be addressed to pathways used in the thermophilic bacterium Petrotoga mobilis, it has genes coding for J.C. ([email protected].) glucosyl-3-phosphoglycerate synthase (GpgS) and mannosylglucosyl-3-phosphoglycerate (MGPG) synthase (MggA). However, unlike Ptg. mobilis, the mesophilic R. baltica uses a novel and very specific MGPG phosphatase (MggB). It also lacks a key enzyme of the alternative pathway in Ptg. mobilis – the mannosylglucosylglycerate synthase (MggS) that catalyses the condensation of glucosylglycerate with GDP-mannose to produce MGG. The R. baltica enzymes GpgS, MggA, and MggB were expressed in E. coli and characterized in terms of kinetic parameters, substrate specificity, temperature and pH dependence. -

And Thermo-Adaptation in Hyperthermophilic Archaea: Identification of Compatible Solutes, Accumulation Profiles, and Biosynthetic Routes in Archaeoglobus Spp
Universidade Nova de Lisboa Osmo- andInstituto thermo de Tecnologia-adaptation Química e Biológica in hyperthermophilic Archaea: Subtitle Subtitle Luís Pedro Gafeira Gonçalves Osmo- and thermo-adaptation in hyperthermophilic Archaea: identification of compatible solutes, accumulation profiles, and biosynthetic routes in Archaeoglobus spp. OH OH OH CDP c c c - CMP O O - PPi O3P P CTP O O O OH OH OH OH OH OH O- C C C O P O O P i Dissertation presented to obtain the Ph.D degree in BiochemistryO O- Instituto de Tecnologia Química e Biológica | Universidade Nova de LisboaP OH O O OH OH OH Oeiras, Luís Pedro Gafeira Gonçalves January, 2008 2008 Universidade Nova de Lisboa Instituto de Tecnologia Química e Biológica Osmo- and thermo-adaptation in hyperthermophilic Archaea: identification of compatible solutes, accumulation profiles, and biosynthetic routes in Archaeoglobus spp. This dissertation was presented to obtain a Ph. D. degree in Biochemistry at the Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa. By Luís Pedro Gafeira Gonçalves Supervised by Prof. Dr. Helena Santos Oeiras, January, 2008 Apoio financeiro da Fundação para a Ciência e Tecnologia (POCI 2010 – Formação Avançada para a Ciência – Medida IV.3) e FSE no âmbito do Quadro Comunitário de apoio, Bolsa de Doutoramento com a referência SFRH / BD / 5076 / 2001. ii ACKNOWNLEDGMENTS The work presented in this thesis, would not have been possible without the help, in terms of time and knowledge, of many people, to whom I am extremely grateful. Firstly and mostly, I need to thank my supervisor, Prof. Helena Santos, for her way of thinking science, her knowledge, her rigorous criticism, and her commitment to science. -

Molecular Evolution of the Oxygen-Binding Hemerythrin Domain
RESEARCH ARTICLE Molecular Evolution of the Oxygen-Binding Hemerythrin Domain Claudia Alvarez-Carreño1, Arturo Becerra1, Antonio Lazcano1,2* 1 Facultad de Ciencias, Universidad Nacional Autónoma de México, Apdo. Postal 70–407, Cd. Universitaria, 04510, Mexico City, Mexico, 2 Miembro de El Colegio Nacional, Ciudad de México, México * [email protected] a11111 Abstract Background The evolution of oxygenic photosynthesis during Precambrian times entailed the diversifica- tion of strategies minimizing reactive oxygen species-associated damage. Four families of OPEN ACCESS oxygen-carrier proteins (hemoglobin, hemerythrin and the two non-homologous families of Citation: Alvarez-Carreño C, Becerra A, Lazcano A arthropodan and molluscan hemocyanins) are known to have evolved independently the (2016) Molecular Evolution of the Oxygen-Binding Hemerythrin Domain. PLoS ONE 11(6): e0157904. capacity to bind oxygen reversibly, providing cells with strategies to cope with the evolution- doi:10.1371/journal.pone.0157904 ary pressure of oxygen accumulation. Oxygen-binding hemerythrin was first studied in Editor: Nikolas Nikolaidis, California State University marine invertebrates but further research has made it clear that it is present in the three Fullerton, UNITED STATES domains of life, strongly suggesting that its origin predated the emergence of eukaryotes. Received: April 5, 2016 Accepted: June 7, 2016 Results Published: June 23, 2016 Oxygen-binding hemerythrins are a monophyletic sub-group of the hemerythrin/HHE (histi- dine, histidine, glutamic acid) cation-binding domain. Oxygen-binding hemerythrin homo- Copyright: © 2016 Alvarez-Carreño et al. This is an open access article distributed under the terms of the logs were unambiguously identified in 367/2236 bacterial, 21/150 archaeal and 4/135 Creative Commons Attribution License, which permits eukaryotic genomes. -

NOC04013 Application.Pdf(PDF, 453
ER-AN-02N 10/02 Application for approval to import into FORM 2N containment any new organism that is not genetically modified, under Section 40 of the Page 1 Hazardous Substances and New Organisms Act 1996 FORM NO2N Application for approval to IMPORT INTO CONTAINMENT ANY NEW ORGANISM THAT IS NOT GENETICALLY MODIFIED under section 40 of the Hazardous Substances and New Organisms Act 1996 Application Title: Importation of sediment/water samples collected from hydrothermal seamounts outside New Zealand Territorial waters containing Extremophilic microorganisms Applicant Organisation: Institute of Geological & Nuclear Sciences ERMA Office use only Application Code: Formally received:____/____/____ ERMA NZ Contact: Initial Fee Paid: $ Application Status: ER-AN-02N 10/02 Application for approval to import into FORM 2N containment any new organism that is not genetically modified, under Section 40 of the Page 2 Hazardous Substances and New Organisms Act 1996 IMPORTANT 1. An associated User Guide is available for this form. You should read the User Guide before completing this form. If you need further guidance in completing this form please contact ERMA New Zealand. 2. This application form covers importation into containment of any new organism that is not genetically modified, under section 40 of the Act. 3. If you are making an application to import into containment a genetically modified organism you should complete Form NO2G, instead of this form (Form NO2N). 4. This form, together with form NO2G, replaces all previous versions of Form 2. Older versions should not now be used. You should periodically check with ERMA New Zealand or on the ERMA New Zealand web site for new versions of this form. -

Marinitoga Lauensis Sp. Nov., a Novel Deep-Sea Hydrothermal Vent Thermophilic Anaerobic Heterotroph with a Prophage
Portland State University PDXScholar Biology Faculty Publications and Presentations Biology 5-1-2019 Marinitoga Lauensis sp. nov., A Novel Deep-sea Hydrothermal Vent Thermophilic Anaerobic Heterotroph with a Prophage Stéphane L'Haridon Univ Brest, CNRS, IFREMER Léna Gouhie CNRS, IFREMER Emily St. John Portland State University, [email protected] Anna-Louise Reysenbach Portland State University, [email protected] Follow this and additional works at: https://pdxscholar.library.pdx.edu/bio_fac Let us know how access to this document benefits ou.y Citation Details Published as: L’Haridon, S., Gouhier, L., John, E. S., & Reysenbach, A. L. (2019). Marinitoga lauensis sp. nov., a novel deep-sea hydrothermal vent thermophilic anaerobic heterotroph with a prophage. Systematic and applied microbiology, 42(3), 343-347. This Post-Print is brought to you for free and open access. It has been accepted for inclusion in Biology Faculty Publications and Presentations by an authorized administrator of PDXScholar. Please contact us if we can make this document more accessible: [email protected]. Accepted Manuscript Title: Marinitoga lauensis sp. nov., a novel deep-sea hydrothermal vent thermophilic anaerobic heterotroph with a prophage Authors: Stephane´ L’Haridon, Lena´ Gouhier, Emily St. John, Anna-Louise Reysenbach PII: S0723-2020(18)30493-4 DOI: https://doi.org/10.1016/j.syapm.2019.02.006 Reference: SYAPM 25981 To appear in: Received date: 30 November 2018 Revised date: 17 February 2019 Accepted date: 26 February 2019 Please cite this article as: Stephane´ L’Haridon, Lena´ Gouhier, Emily St.John, Anna- Louise Reysenbach, Marinitoga lauensis sp.nov., a novel deep-sea hydrothermal vent thermophilic anaerobic heterotroph with a prophage, Systematic and Applied Microbiology https://doi.org/10.1016/j.syapm.2019.02.006 This is a PDF file of an unedited manuscript that has been accepted for publication. -

Distribution of Aerobic Arsenite Oxidase Genes Within the Aquificales
Interdisciplinary Studies on Environmental Chemistry — Biological Responses to Contaminants, Eds., N. Hamamura, S. Suzuki, S. Mendo, C. M. Barroso, H. Iwata and S. Tanabe, pp. 47–55. © by TERRAPUB, 2010. Distribution of Aerobic Arsenite Oxidase Genes within the Aquificales Natsuko HAMAMURA1,2, Rich E. MACUR3, Yitai LIU2, William P. INSKEEP3 and Anna-Louise REYSENBACH2 1Center for Marine Environmental Studies, Ehime University, Matsuyama 790-8577 Japan 2Department of Biology, Portland State University, Portland, OR 97201 U.S.A. 3Thermal Biology Institute and Department of Land Resources and Environmental Sciences, Montana State University, Bozeman, MT 59717 U.S.A. (Received 4 January 2010; accepted 25 January 2010) Abstract—The Aquificales are one of the dominant bacterial orders associated with arsenic-rich geothermal environments, however, their role in arsenic transformations has not been fully characterized. In this report, we examined the distribution of aerobic arsenite oxidase genes (aroA-like) among 26 Aquificales isolates from geographically distinct marine and terrestrial hydrothermal systems. Arsenite-oxidase genes were detected in two Hydrogenobacter strains and one Sulfurihydrogenibium strain isolated from terrestrial springs in the Uzon Caldera and the Geyser Valley of Kamchatka, Russia, but were absent in other phylogenetically closely related strains isolated from the same thermal systems. The arsenite-oxidation activities of these aroA-positive strains were also confirmed. The newly identified aroA- like sequences share high sequence similarity to aroA genes identified in other thermophilic bacteria, but still formed separate clades from previously identified aroA sequences. These results extend our knowledge of the diversity and distribution of Mo-pterin arsenite oxidases in the Aquificales, an important step in understanding the evolutionary relationships of deeply-rooted bacterial arsenite oxidases relative to the diversity of aroA-like genes now known to exist across the bacterial domain. -

Alpha-Carbonic Anhydrases from Hydrothermal Vent Sources As Potential Carbon Dioxide Sequestration Agents: in Silico Sequence, Structure and Dynamics Analyses
International Journal of Molecular Sciences Article Alpha-Carbonic Anhydrases from Hydrothermal Vent Sources as Potential Carbon Dioxide Sequestration Agents: In Silico Sequence, Structure and Dynamics Analyses Colleen Varaidzo Manyumwa 1 , Reza Zolfaghari Emameh 2 and Özlem Tastan Bishop 1,* 1 Research Unit in Bioinformatics (RUBi), Department of Biochemistry and Microbiology, Rhodes University, Makhanda/Grahamstown 6140, South Africa; [email protected] 2 Department of Energy and Environmental Biotechnology, National Institute of Genetic Engineering and Biotechnology (NIGEB), Tehran 14965/161, Iran; [email protected] * Correspondence: [email protected]; Tel.: +27-46-603-8072; Fax: +27-46-603-7576 Received: 28 September 2020; Accepted: 27 October 2020; Published: 29 October 2020 Abstract: With the increase in CO2 emissions worldwide and its dire effects, there is a need to reduce CO2 concentrations in the atmosphere. Alpha-carbonic anhydrases (α-CAs) have been identified as suitable sequestration agents. This study reports the sequence and structural analysis of 15 α-CAs from bacteria, originating from hydrothermal vent systems. Structural analysis of the multimers enabled the identification of hotspot and interface residues. Molecular dynamics simulations of the homo-multimers were performed at 300 K, 363 K, 393 K and 423 K to unearth potentially thermostable α-CAs. Average betweenness centrality (BC) calculations confirmed the relevance of some hotspot and interface residues. The key residues responsible for dimer thermostability were identified by comparing fluctuating interfaces with stable ones, and were part of conserved motifs. Crucial long-lived hydrogen bond networks were observed around residues with high BC values. Dynamic cross correlation fortified the relevance of oligomerization of these proteins, thus the importance of simulating them in their multimeric forms. -

Thermostable Rnase P Rnas Lacking P18 Identified in the Aquificales
JOBNAME: RNA 12#11 2006 PAGE: 1 OUTPUT: Wednesday September 27 16:21:46 2006 csh/RNA/125782/rna2428 Downloaded from rnajournal.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press REPORT Thermostable RNase P RNAs lacking P18 identified in the Aquificales MICHAL MARSZALKOWSKI,1 JAN-HENDRIK TEUNE,2 GERHARD STEGER,2 ROLAND K. HARTMANN,1 and DAGMAR K. WILLKOMM1 1Philipps-Universita¨t Marburg, Institut fu¨r Pharmazeutische Chemie, D-35037 Marburg, Germany 2Heinrich-Heine-Universita¨tDu¨sseldorf, Institut fu¨r Physikalische Biologie, D-40225 Du¨sseldorf, Germany ABSTRACT The RNase P RNA (rnpB) and protein (rnpA) genes were identified in the two Aquificales Sulfurihydrogenibium azorense and Persephonella marina. In contrast, neither of the two genes has been found in the sequenced genome of their close relative, Aquifex aeolicus. As in most bacteria, the rnpA genes of S. azorense and P. marina are preceded by the rpmH gene coding for ribosomal protein L34. This genetic region, including several genes up- and downstream of rpmH, is uniquely conserved among all three Aquificales strains, except that rnpA is missing in A. aeolicus. The RNase P RNAs (P RNAs) of S. azorense and P. marina are active catalysts that can be activated by heterologous bacterial P proteins at low salt. Although the two P RNAs lack helix P18 and thus one of the three major interdomain tertiary contacts, they are more thermostable than Escherichia coli P RNA and require higher temperatures for proper folding. Related to their thermostability, both RNAs include a subset of structural idiosyncrasies in their S domains, which were recently demonstrated to determine the folding properties of the thermostable S domain of Thermus thermophilus P RNA. -

Electron Donors and Acceptors for Members of the Family Beggiatoaceae
Electron donors and acceptors for members of the family Beggiatoaceae Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften - Dr. rer. nat. - dem Fachbereich Biologie/Chemie der Universit¨at Bremen vorgelegt von Anne-Christin Kreutzmann aus Hildesheim Bremen, November 2013 Die vorliegende Doktorarbeit wurde in der Zeit von Februar 2009 bis November 2013 am Max-Planck-Institut f¨ur marine Mikrobiologie in Bremen angefertigt. 1. Gutachterin: Prof. Dr. Heide N. Schulz-Vogt 2. Gutachter: Prof. Dr. Ulrich Fischer 3. Pr¨uferin: Prof. Dr. Nicole Dubilier 4. Pr¨ufer: Dr. Timothy G. Ferdelman Tag des Promotionskolloquiums: 16.12.2013 To Finn Summary The family Beggiatoaceae comprises large, colorless sulfur bacteria, which are best known for their chemolithotrophic metabolism, in particular the oxidation of re- duced sulfur compounds with oxygen or nitrate. This thesis contributes to a more comprehensive understanding of the physiology and ecology of these organisms with several studies on different aspects of their dissimilatory metabolism. Even though the importance of inorganic sulfur substrates as electron donors for the Beggiatoaceae has long been recognized, it was not possible to derive a general model of sulfur compound oxidation in this family, owing to the fact that most of its members can currently not be cultured. Such a model has now been developed by integrating information from six Beggiatoaceae draft genomes with available literature data (Section 2). This model proposes common metabolic pathways of sulfur compound oxidation and evaluates whether the involved enzymes are likely to be of ancestral origin for the family. In Section 3 the sulfur metabolism of the Beggiatoaceae is explored from a dif- ferent perspective. -

Carbon Isotopic Fractionations Associated with Thermophilic Bacteria Thermotoga Maritima and Persephonella Marina
Environmental Microbiology (2002) 4(1), 58–64 Carbon isotopic fractionations associated with thermophilic bacteria Thermotoga maritima and Persephonella marina 1 1 Chuanlun L. Zhang, * Qi Ye, Anna-Louise pathway may be used for CO2 fixation. This is sup- Reysenbach,2 Dorothee Götz,2 Aaron Peacock,3 ported by small fractionation between biomass and 3 4 4 David C. White, Juske Horita, David R. Cole, CO2 (e = -3.8‰ to -5.0‰), which is similar to frac- Jon Fong,5 Lisa Pratt,5 Jiasong Fang6 tionations reported for other organisms using similar 7 and Yongsong Huang CO2 fixation pathways. Identification of the exact 1Department of Geological Sciences, pathway will require biochemical assay for specific University of Missouri, Columbia, MO 65211, enzymes associated with the reversed TCA cycle or USA. the 3-hydroxypropionate pathway. 2Department of Biology, Portland State University, Portland, OR 97201, USA. Introduction 3Institute for Applied Microbiology, The University of Tennessee, Knoxville, TN 37932–2575, USA. In situ microbial production is an important source of 4Chemical and Analytical Sciences Division, organic matter in hydrothermal vents (McCollom and Oak Ridge National Laboratory, Oak Ridge, Shock, 1997), yet the mechanisms of carbon cycling by TN 37831–6110, USA. vent microorganisms, especially by thermophiles, are 5Department of Geological Sciences, Indiana University, poorly understood. Isotopic fractionations of elements Bloomington, IN 47405, USA. such as carbon have traditionally been used to differ- 6Department of Civil and Environmental Engineering, entiate between biotic and abiotic processes because University of Michigan, Ann Arbor, MI 48109–2099, biologically mediated chemical reactions preferentially 12 13 USA. incorporate lighter isotopes (i.e.