Interactions of Aquificae with Mercury And
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Genomics 98 (2011) 370–375
Genomics 98 (2011) 370–375 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno Whole-genome comparison clarifies close phylogenetic relationships between the phyla Dictyoglomi and Thermotogae Hiromi Nishida a,⁎, Teruhiko Beppu b, Kenji Ueda b a Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan b Life Science Research Center, College of Bioresource Sciences, Nihon University, Fujisawa, Japan article info abstract Article history: The anaerobic thermophilic bacterial genus Dictyoglomus is characterized by the ability to produce useful Received 2 June 2011 enzymes such as amylase, mannanase, and xylanase. Despite the significance, the phylogenetic position of Accepted 1 August 2011 Dictyoglomus has not yet been clarified, since it exhibits ambiguous phylogenetic positions in a single gene Available online 7 August 2011 sequence comparison-based analysis. The number of substitutions at the diverging point of Dictyoglomus is insufficient to show the relationships in a single gene comparison-based analysis. Hence, we studied its Keywords: evolutionary trait based on whole-genome comparison. Both gene content and orthologous protein sequence Whole-genome comparison Dictyoglomus comparisons indicated that Dictyoglomus is most closely related to the phylum Thermotogae and it forms a Bacterial systematics monophyletic group with Coprothermobacter proteolyticus (a constituent of the phylum Firmicutes) and Coprothermobacter proteolyticus Thermotogae. Our findings indicate that C. proteolyticus does not belong to the phylum Firmicutes and that the Thermotogae phylum Dictyoglomi is not closely related to either the phylum Firmicutes or Synergistetes but to the phylum Thermotogae. © 2011 Elsevier Inc. -

Diversity of Understudied Archaeal and Bacterial Populations of Yellowstone National Park: from Genes to Genomes Daniel Colman
University of New Mexico UNM Digital Repository Biology ETDs Electronic Theses and Dissertations 7-1-2015 Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes Daniel Colman Follow this and additional works at: https://digitalrepository.unm.edu/biol_etds Recommended Citation Colman, Daniel. "Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes." (2015). https://digitalrepository.unm.edu/biol_etds/18 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biology ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Daniel Robert Colman Candidate Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Cristina Takacs-Vesbach , Chairperson Robert Sinsabaugh Laura Crossey Diana Northup i Diversity of understudied archaeal and bacterial populations from Yellowstone National Park: from genes to genomes by Daniel Robert Colman B.S. Biology, University of New Mexico, 2009 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biology The University of New Mexico Albuquerque, New Mexico July 2015 ii DEDICATION I would like to dedicate this dissertation to my late grandfather, Kenneth Leo Colman, associate professor of Animal Science in the Wool laboratory at Montana State University, who even very near the end of his earthly tenure, thought it pertinent to quiz my knowledge of oxidized nitrogen compounds. He was a man of great curiosity about the natural world, and to whom I owe an acknowledgement for his legacy of intellectual (and actual) wanderlust. -
A Deep Learning Approach to Pattern Recognition for Short DNA Sequences”
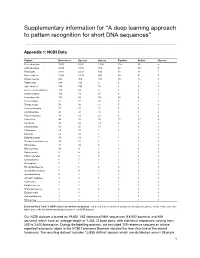
Supplementary information for “A deep learning approach to pattern recognition for short DNA sequences” Appendix 1: NCBI Data Phylum References Species Genera Families Orders Classes Proteobacteria 7,053 5,061 1,106 158 55 9 Actinobacteria 4,768 3,313 383 68 29 6 Firmicutes 3,814 2,531 499 56 13 7 Bacteroidetes 1,934 1,525 360 39 8 7 Euryarchaeota 834 450 100 28 13 8 Tenericutes 266 195 8 5 4 1 Spirochaetes 146 100 16 6 4 1 Deinococcus-Thermus 118 99 9 3 2 1 Crenarchaeota 113 61 27 8 5 1 Cyanobacteria 113 88 59 30 8 2 Fusobacteria 75 37 10 2 1 1 Thermotogae 70 48 13 5 4 1 Verrucomicrobia 57 53 22 7 4 3 Acidobacteria 45 41 19 5 5 4 Planctomycetes 44 31 23 5 3 2 Chloroflexi 44 35 26 15 12 8 Aquificae 43 32 14 4 2 1 Synergistetes 31 25 15 1 1 1 Chlamydiae 28 18 7 5 2 1 Chlorobi 21 16 5 1 1 1 Deferribacteres 15 11 7 1 1 1 Thermodesulfobacteria 14 12 5 1 1 1 Nitrospirae 11 10 3 1 1 1 Fibrobacteres 10 4 3 3 3 3 Balneolaeota 9 9 4 1 1 1 Chrysiogenetes 6 4 3 1 1 1 Lentisphaerae 6 5 3 3 3 2 Dictyoglomi 5 2 1 1 1 1 Rhodothermaeota 5 5 3 2 1 1 Gemmatimonadetes 5 4 3 2 2 2 Ignavibacteriae 4 2 2 2 1 1 Armatimonadetes 4 3 3 3 3 3 Caldiserica 3 1 1 1 1 1 Calditrichaeota 2 2 1 1 1 1 Thaumarchaeota 2 2 2 2 2 2 Elusimicrobia 2 1 1 1 1 1 Kiritimatiellaeota 1 1 1 1 1 1 Nitrospinae 1 1 1 1 1 1 Extended Data Table 1: NCBI dataset breakdown by phylum. -

Cone-Forming Chloroflexi Mats As Analogs of Conical
268 Appendix 2 CONE-FORMING CHLOROFLEXI MATS AS ANALOGS OF CONICAL STROMATOLITE FORMATION WITHOUT CYANOBACTERIA Lewis M. Ward, Woodward W. Fischer, Katsumi Matsuura, and Shawn E. McGlynn. In preparation. Abstract Modern microbial mats provide useful process analogs for understanding the mechanics behind the production of ancient stromatolites. However, studies to date have focused on mats composed predominantly of oxygenic Cyanobacteria (Oxyphotobacteria) and algae, which makes it difficult to assess a unique role of oxygenic photosynthesis in stromatolite morphogenesis, versus different mechanics such as phototaxis and filamentous growth. Here, we characterize Chloroflexi-rich hot spring microbial mats from Nakabusa Onsen, Nagano Prefecture, Japan. This spring supports cone-forming microbial mats in both upstream high-temperature, sulfidic regions dominated by filamentous anoxygenic phototrophic Chloroflexi, as well as downstream Cyanobacteria-dominated mats. These mats produce similar morphologies analogous to conical stromatolites despite metabolically and taxonomically divergent microbial communities as revealed by 16S and shotgun metagenomic sequencing and microscopy. These data illustrate that anoxygenic filamentous microorganisms appear to be capable of producing similar mat morphologies as those seen in Oxyphotobacteria-dominated systems and commonly associated with 269 conical Precambrian stromatolites, and that the processes leading to the development of these features is more closely related with characteristics such as hydrology and cell morphology and motility. Introduction Stromatolites are “attached, lithified sedimentary growth structures, accretionary away from a point or limited surface of initiation” (Grotzinger and Knoll 1999). Behind this description lies a wealth of sedimentary structures with a record dating back over 3.7 billion years that may be one of the earliest indicators of life on Earth (Awramik 1992, Nutman et al. -

Genomic Analysis of Family UBA6911 (Group 18 Acidobacteria)
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 Genomic analysis of family UBA6911 (Group 18 3 Acidobacteria) expands the metabolic capacities of the 4 phylum and highlights adaptations to terrestrial habitats. 5 6 Archana Yadav1, Jenna C. Borrelli1, Mostafa S. Elshahed1, and Noha H. Youssef1* 7 8 1Department of Microbiology and Molecular Genetics, Oklahoma State University, Stillwater, 9 OK 10 *Correspondence: Noha H. Youssef: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 11 Abstract 12 Approaches for recovering and analyzing genomes belonging to novel, hitherto unexplored 13 bacterial lineages have provided invaluable insights into the metabolic capabilities and 14 ecological roles of yet-uncultured taxa. The phylum Acidobacteria is one of the most prevalent 15 and ecologically successful lineages on earth yet, currently, multiple lineages within this phylum 16 remain unexplored. Here, we utilize genomes recovered from Zodletone spring, an anaerobic 17 sulfide and sulfur-rich spring in southwestern Oklahoma, as well as from multiple disparate soil 18 and non-soil habitats, to examine the metabolic capabilities and ecological role of members of 19 the family UBA6911 (group18) Acidobacteria. -

Table S4. Phylogenetic Distribution of Bacterial and Archaea Genomes in Groups A, B, C, D, and X
Table S4. Phylogenetic distribution of bacterial and archaea genomes in groups A, B, C, D, and X. Group A a: Total number of genomes in the taxon b: Number of group A genomes in the taxon c: Percentage of group A genomes in the taxon a b c cellular organisms 5007 2974 59.4 |__ Bacteria 4769 2935 61.5 | |__ Proteobacteria 1854 1570 84.7 | | |__ Gammaproteobacteria 711 631 88.7 | | | |__ Enterobacterales 112 97 86.6 | | | | |__ Enterobacteriaceae 41 32 78.0 | | | | | |__ unclassified Enterobacteriaceae 13 7 53.8 | | | | |__ Erwiniaceae 30 28 93.3 | | | | | |__ Erwinia 10 10 100.0 | | | | | |__ Buchnera 8 8 100.0 | | | | | | |__ Buchnera aphidicola 8 8 100.0 | | | | | |__ Pantoea 8 8 100.0 | | | | |__ Yersiniaceae 14 14 100.0 | | | | | |__ Serratia 8 8 100.0 | | | | |__ Morganellaceae 13 10 76.9 | | | | |__ Pectobacteriaceae 8 8 100.0 | | | |__ Alteromonadales 94 94 100.0 | | | | |__ Alteromonadaceae 34 34 100.0 | | | | | |__ Marinobacter 12 12 100.0 | | | | |__ Shewanellaceae 17 17 100.0 | | | | | |__ Shewanella 17 17 100.0 | | | | |__ Pseudoalteromonadaceae 16 16 100.0 | | | | | |__ Pseudoalteromonas 15 15 100.0 | | | | |__ Idiomarinaceae 9 9 100.0 | | | | | |__ Idiomarina 9 9 100.0 | | | | |__ Colwelliaceae 6 6 100.0 | | | |__ Pseudomonadales 81 81 100.0 | | | | |__ Moraxellaceae 41 41 100.0 | | | | | |__ Acinetobacter 25 25 100.0 | | | | | |__ Psychrobacter 8 8 100.0 | | | | | |__ Moraxella 6 6 100.0 | | | | |__ Pseudomonadaceae 40 40 100.0 | | | | | |__ Pseudomonas 38 38 100.0 | | | |__ Oceanospirillales 73 72 98.6 | | | | |__ Oceanospirillaceae -

Evolution of the 3-Hydroxypropionate Bicycle and Recent Transfer of Anoxygenic Photosynthesis Into the Chloroflexi
Evolution of the 3-hydroxypropionate bicycle and recent transfer of anoxygenic photosynthesis into the Chloroflexi Patrick M. Shiha,b,1, Lewis M. Wardc, and Woodward W. Fischerc,1 aFeedstocks Division, Joint BioEnergy Institute, Emeryville, CA 94608; bEnvironmental Genomics and Systems Biology Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720; and cDivision of Geological and Planetary Sciences, California Institute of Technology, Pasadena, CA 91125 Edited by Bob B. Buchanan, University of California, Berkeley, CA, and approved August 21, 2017 (received for review June 14, 2017) Various lines of evidence from both comparative biology and the provide a hard geological constraint on these analyses, the timing geologic record make it clear that the biochemical machinery for of these evolutionary events remains relative, thus highlighting anoxygenic photosynthesis was present on early Earth and provided the uncertainty in our understanding of when and how anoxy- the evolutionary stock from which oxygenic photosynthesis evolved genic photosynthesis may have originated. ca. 2.3 billion years ago. However, the taxonomic identity of these A less recognized alternative is that anoxygenic photosynthesis early anoxygenic phototrophs is uncertain, including whether or not might have been acquired in modern bacterial clades relatively they remain extant. Several phototrophic bacterial clades are thought recently. This possibility is supported by the observation that to have evolved before oxygenic photosynthesis emerged, including anoxygenic photosynthesis often sits within a derived position in the Chloroflexi, a phylum common across a wide range of modern the phyla in which it is found (3). Moreover, it is increasingly environments. Although Chloroflexi have traditionally been thought being recognized that horizontal gene transfer (HGT) has likely to be an ancient phototrophic lineage, genomics has revealed a much played a major role in the distribution of phototrophy (8–10). -

7. References
University of Akureyri Department of Natural Resource Science 7. References Ammann, E. C., Reed, L. L., & Durichek, J. J. (1968). Gas consumptions and Growth Rate of Hydrogenomonas eutropha in Continuous Culture. Applied Microbiology , 16, (6), 822-826. Aguiar, P., Beveridge, T. J., & Reysenbach, A.-L. (2004). Sulfurihydrogenibium azorense, sp. nov., a thermophilic hydrogen oxidizing microaerophile from terrestrial hot springs in the Azores. International Journal of Systematic and Evolutionary Microbiology , 54, 33-39. Altschul, S., Gish, W., Miller, W., Myers, E., & Lipman, D. (1990). "Basic local alignment search tool.". J. Mol. Biol. , 215:403-410. Amend, J., & Shock, E. (2001). Energetics of overall metabolic reactions of thermophilic and hyperthermophilic Archaea and Bacteria. FEMS Microbiology Reviews , 25, 175-243. Aragno, M. (1978). Enrichment, isolation and preliminary characterization of a thermophilic, endospore-forming hydrogen bacterium. FEMS Micobiol. Lett. , 3: 13-15. Aragno, M. (1992). The Thermophilic, Aerobic, Hydrogen-Oxidizing (Knallgas) Bacteria. In A. Balows, H. Trüper, M. Dworkin, W. Harder, & K. Schleifer, The Prokaryotes, a handbook on biology of bacteria. 2nd ed. vol. 4 (pp. 3917-3933.). New York: Springer Verlag. Aragno, M., & Schlegel, H. G. (1992). The mesophilic Hydrogen-Oxidizing (Knallgas) Bacteria. In A. Balows, H. Truper, M. Dworkin, W. Harder, & K.-H. Schleifer, The Prokaryotes 2nd. ed. (pp. 344-384). New York: Springer. Ármannson, H. (2002, May 30-31). Erindi á ráðstefnu um málefni veitufyrirtækja . Grænt bókhald í jarðhita- samanburður á útblæstri við aðra orkugjafa . Akureyri, Iceland: Samorka. Bae, S., Kwak, K., Kim, S., Chung, S., & Igarashi, Y. (2001). Isolation and Characterization of CO2-Fixing Hydrogen -Oxidizing Marine 109 University of Akureyri Department of Natural Resource Science Bacteria. -

Yu-Chen Ling and John W. Moreau
Microbial Distribution and Activity in a Coastal Acid Sulfate Soil System Introduction: Bioremediation in Yu-Chen Ling and John W. Moreau coastal acid sulfate soil systems Method A Coastal acid sulfate soil (CASS) systems were School of Earth Sciences, University of Melbourne, Melbourne, VIC 3010, Australia formed when people drained the coastal area Microbial distribution controlled by environmental parameters Microbial activity showed two patterns exposing the soil to the air. Drainage makes iron Microbial structures can be grouped into three zones based on the highest similarity between samples (Fig. 4). Abundant populations, such as Deltaproteobacteria, kept constant activity across tidal cycling, whereas rare sulfides oxidize and release acidity to the These three zones were consistent with their geological background (Fig. 5). Zone 1: Organic horizon, had the populations changed activity response to environmental variations. Activity = cDNA/DNA environment, low pH pore water further dissolved lowest pH value. Zone 2: surface tidal zone, was influenced the most by tidal activity. Zone 3: Sulfuric zone, Abundant populations: the heavy metals. The acidity and toxic metals then Method A Deltaproteobacteria Deltaproteobacteria this area got neutralized the most. contaminate coastal and nearby ecosystems and Method B 1.5 cause environmental problems, such as fish kills, 1.5 decreased rice yields, release of greenhouse gases, Chloroflexi and construction damage. In Australia, there is Gammaproteobacteria Gammaproteobacteria about a $10 billion “legacy” from acid sulfate soils, Chloroflexi even though Australia is only occupied by around 1.0 1.0 Cyanobacteria,@ Acidobacteria Acidobacteria Alphaproteobacteria 18% of the global acid sulfate soils. Chloroplast Zetaproteobacteria Rare populations: Alphaproteobacteria Method A log(RNA(%)+1) Zetaproteobacteria log(RNA(%)+1) Method C Method B 0.5 0.5 Cyanobacteria,@ Bacteroidetes Chloroplast Firmicutes Firmicutes Bacteroidetes Planctomycetes Planctomycetes Ac8nobacteria Fig. -

Distribution of Arsenite-Oxidizing Bacteria and Its Correlation with Environmental Factors in Geothermal Areas of Tengchong, Yunnan, China
E3S Web of Conferences 98, 02002 (2019) https://doi.org/10.1051/e3sconf/20199802002 WRI-16 Distribution of arsenite-oxidizing bacteria and its correlation with environmental factors in geothermal areas of Tengchong, Yunnan, China Ping Li1,*, Dawei Jiang1, and Zhou Jiang1,2 1State Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences, Wuhan, China 2School of Environmental Studies, China University of Geosciences, Wuhan, China Abstract. Arsenic (As) is an ubiquitous constituent in geothermal water. Arsenite (AsIII) is oxidized via microbial processes as the waters equilibrate with oxygen in the geothermal effluent. The distribution of arsenite oxdizing bacteria and its correlation with environment factors were studied in Tengchong geothermal areas of Yunnan, China. A total of 230 aioA clone sequences were obtained and these sequences were affiliated with four phyla: Betaproteobacteria, Alphaproteobacteria, Deinococcus- Thermus and Aquificae. Temperature was negatively correlated with aioA diversity and was the only environment factor that had correlation with diversity index. Betaproteobacteria was mainly distributed in low temperature (T = 28 to 43 oC) and circumneutral or light alkaline (pH = 7 to 9) springs; Alphaproteobacteria was mainly predominant in low pH (pH = 3.3 to 3.6) springs; Deinococcus-Thermus and Aquificae mainly inhabited in high temperature (T=55 to 78 oC) springs with a wide range of pH. Usually, Deinococcus-Thermus was dominant when springs had a pH within 4.0 to 8.0. Aquificae was dominated in springs with pH > 8.0 or pH < 4.0. 1 Diversity of aioA gene A total of 230 aioA gene clone sequences from 10 sample sites were subjected to sequence similarity analysis. -

MIAMI UNIVERSITY the Graduate School Certificate for Approving The
MIAMI UNIVERSITY The Graduate School Certificate for Approving the Dissertation We hereby approve the Dissertation of Qiuyuan Huang Candidate for the Degree: Doctor of Philosophy _______________________________________ Hailiang Dong, Director ________________________________________ Yildirim Dilek, Reader ________________________________________ Jonathan Levy, Reader ______________________________________ Chuanlun Zhang, External examiner ______________________________________ Annette Bollmann, Graduate School Representative ABSTRACT GEOMICROBIAL INVESTIGATIONS ON EXTREME ENVIRONMENTS: LINKING GEOCHEMISTRY TO MICROBIAL ECOLOGY IN TERRESTRIAL HOT SPRINGS AND SALINE LAKES by Qiuyuan Huang Terrestrial hot springs and saline lakes represent two extreme environments for microbial life and constitute an important part of global ecosystems that affect the biogeochemical cycling of life-essential elements. Despite the advances in our understanding of microbial ecology in the past decade, important questions remain regarding the link between microbial diversity and geochemical factors under these extreme conditions. This dissertation first investigates a series of hot springs with wide ranges of temperature (26-92oC) and pH (3.72-8.2) from the Tibetan Plateau in China and the Philippines. Within each region, microbial diversity and geochemical conditions were studied using an integrated approach with 16S rRNA molecular phylogeny and a suite of geochemical analyses. In Tibetan springs, the microbial community was dominated by archaeal phylum Thaumarchaeota -

Characterization of the Uncommon Enzymes from (2004)
OPEN Mannosylglucosylglycerate biosynthesis SUBJECT AREAS: in the deep-branching phylum WATER MICROBIOLOGY MARINE MICROBIOLOGY Planctomycetes: characterization of the HOMEOSTASIS MULTIENZYME COMPLEXES uncommon enzymes from Rhodopirellula Received baltica 13 March 2013 Sofia Cunha1, Ana Filipa d’Avo´1, Ana Mingote2, Pedro Lamosa3, Milton S. da Costa1,4 & Joana Costa1,4 Accepted 23 July 2013 1Center for Neuroscience and Cell Biology, University of Coimbra, 3004-517 Coimbra, Portugal, 2Instituto de Tecnologia Quı´mica Published e Biolo´gica, Universidade Nova de Lisboa, 2780-157 Oeiras, Portugal, 3Centro de Ressonaˆncia Magne´tica Anto´nio Xavier, 7 August 2013 Instituto de Tecnologia Quı´mica e Biolo´gica, Universidade Nova de Lisboa, 2781-901 Oeiras, Portugal, 4Department of Life Sciences, University of Coimbra, Apartado 3046, 3001-401 Coimbra, Portugal. Correspondence and The biosynthetic pathway for the rare compatible solute mannosylglucosylglycerate (MGG) accumulated by requests for materials Rhodopirellula baltica, a marine member of the phylum Planctomycetes, has been elucidated. Like one of the should be addressed to pathways used in the thermophilic bacterium Petrotoga mobilis, it has genes coding for J.C. ([email protected].) glucosyl-3-phosphoglycerate synthase (GpgS) and mannosylglucosyl-3-phosphoglycerate (MGPG) synthase (MggA). However, unlike Ptg. mobilis, the mesophilic R. baltica uses a novel and very specific MGPG phosphatase (MggB). It also lacks a key enzyme of the alternative pathway in Ptg. mobilis – the mannosylglucosylglycerate synthase (MggS) that catalyses the condensation of glucosylglycerate with GDP-mannose to produce MGG. The R. baltica enzymes GpgS, MggA, and MggB were expressed in E. coli and characterized in terms of kinetic parameters, substrate specificity, temperature and pH dependence.