Alpha-Carbonic Anhydrases from Hydrothermal Vent Sources As Potential Carbon Dioxide Sequestration Agents: in Silico Sequence, Structure and Dynamics Analyses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Understudied Archaeal and Bacterial Populations of Yellowstone National Park: from Genes to Genomes Daniel Colman
University of New Mexico UNM Digital Repository Biology ETDs Electronic Theses and Dissertations 7-1-2015 Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes Daniel Colman Follow this and additional works at: https://digitalrepository.unm.edu/biol_etds Recommended Citation Colman, Daniel. "Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes." (2015). https://digitalrepository.unm.edu/biol_etds/18 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biology ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Daniel Robert Colman Candidate Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Cristina Takacs-Vesbach , Chairperson Robert Sinsabaugh Laura Crossey Diana Northup i Diversity of understudied archaeal and bacterial populations from Yellowstone National Park: from genes to genomes by Daniel Robert Colman B.S. Biology, University of New Mexico, 2009 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biology The University of New Mexico Albuquerque, New Mexico July 2015 ii DEDICATION I would like to dedicate this dissertation to my late grandfather, Kenneth Leo Colman, associate professor of Animal Science in the Wool laboratory at Montana State University, who even very near the end of his earthly tenure, thought it pertinent to quiz my knowledge of oxidized nitrogen compounds. He was a man of great curiosity about the natural world, and to whom I owe an acknowledgement for his legacy of intellectual (and actual) wanderlust. -
A Deep Learning Approach to Pattern Recognition for Short DNA Sequences”
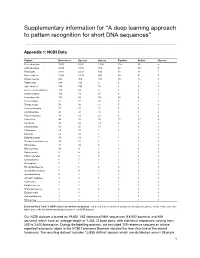
Supplementary information for “A deep learning approach to pattern recognition for short DNA sequences” Appendix 1: NCBI Data Phylum References Species Genera Families Orders Classes Proteobacteria 7,053 5,061 1,106 158 55 9 Actinobacteria 4,768 3,313 383 68 29 6 Firmicutes 3,814 2,531 499 56 13 7 Bacteroidetes 1,934 1,525 360 39 8 7 Euryarchaeota 834 450 100 28 13 8 Tenericutes 266 195 8 5 4 1 Spirochaetes 146 100 16 6 4 1 Deinococcus-Thermus 118 99 9 3 2 1 Crenarchaeota 113 61 27 8 5 1 Cyanobacteria 113 88 59 30 8 2 Fusobacteria 75 37 10 2 1 1 Thermotogae 70 48 13 5 4 1 Verrucomicrobia 57 53 22 7 4 3 Acidobacteria 45 41 19 5 5 4 Planctomycetes 44 31 23 5 3 2 Chloroflexi 44 35 26 15 12 8 Aquificae 43 32 14 4 2 1 Synergistetes 31 25 15 1 1 1 Chlamydiae 28 18 7 5 2 1 Chlorobi 21 16 5 1 1 1 Deferribacteres 15 11 7 1 1 1 Thermodesulfobacteria 14 12 5 1 1 1 Nitrospirae 11 10 3 1 1 1 Fibrobacteres 10 4 3 3 3 3 Balneolaeota 9 9 4 1 1 1 Chrysiogenetes 6 4 3 1 1 1 Lentisphaerae 6 5 3 3 3 2 Dictyoglomi 5 2 1 1 1 1 Rhodothermaeota 5 5 3 2 1 1 Gemmatimonadetes 5 4 3 2 2 2 Ignavibacteriae 4 2 2 2 1 1 Armatimonadetes 4 3 3 3 3 3 Caldiserica 3 1 1 1 1 1 Calditrichaeota 2 2 1 1 1 1 Thaumarchaeota 2 2 2 2 2 2 Elusimicrobia 2 1 1 1 1 1 Kiritimatiellaeota 1 1 1 1 1 1 Nitrospinae 1 1 1 1 1 1 Extended Data Table 1: NCBI dataset breakdown by phylum. -

Thermophilic Lithotrophy and Phototrophy in an Intertidal, Iron-Rich, Geothermal Spring 2 3 Lewis M
bioRxiv preprint doi: https://doi.org/10.1101/428698; this version posted September 27, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Thermophilic Lithotrophy and Phototrophy in an Intertidal, Iron-rich, Geothermal Spring 2 3 Lewis M. Ward1,2,3*, Airi Idei4, Mayuko Nakagawa2,5, Yuichiro Ueno2,5,6, Woodward W. 4 Fischer3, Shawn E. McGlynn2* 5 6 1. Department of Earth and Planetary Sciences, Harvard University, Cambridge, MA 02138 USA 7 2. Earth-Life Science Institute, Tokyo Institute of Technology, Meguro, Tokyo, 152-8550, Japan 8 3. Division of Geological and Planetary Sciences, California Institute of Technology, Pasadena, CA 9 91125 USA 10 4. Department of Biological Sciences, Tokyo Metropolitan University, Hachioji, Tokyo 192-0397, 11 Japan 12 5. Department of Earth and Planetary Sciences, Tokyo Institute of Technology, Meguro, Tokyo, 13 152-8551, Japan 14 6. Department of Subsurface Geobiological Analysis and Research, Japan Agency for Marine-Earth 15 Science and Technology, Natsushima-cho, Yokosuka 237-0061, Japan 16 Correspondence: [email protected] or [email protected] 17 18 Abstract 19 Hydrothermal systems, including terrestrial hot springs, contain diverse and systematic 20 arrays of geochemical conditions that vary over short spatial scales due to progressive interaction 21 between the reducing hydrothermal fluids, the oxygenated atmosphere, and in some cases 22 seawater. At Jinata Onsen, on Shikinejima Island, Japan, an intertidal, anoxic, iron- and 23 hydrogen-rich hot spring mixes with the oxygenated atmosphere and sulfate-rich seawater over 24 short spatial scales, creating an enormous range of redox environments over a distance ~10 m. -

7. References
University of Akureyri Department of Natural Resource Science 7. References Ammann, E. C., Reed, L. L., & Durichek, J. J. (1968). Gas consumptions and Growth Rate of Hydrogenomonas eutropha in Continuous Culture. Applied Microbiology , 16, (6), 822-826. Aguiar, P., Beveridge, T. J., & Reysenbach, A.-L. (2004). Sulfurihydrogenibium azorense, sp. nov., a thermophilic hydrogen oxidizing microaerophile from terrestrial hot springs in the Azores. International Journal of Systematic and Evolutionary Microbiology , 54, 33-39. Altschul, S., Gish, W., Miller, W., Myers, E., & Lipman, D. (1990). "Basic local alignment search tool.". J. Mol. Biol. , 215:403-410. Amend, J., & Shock, E. (2001). Energetics of overall metabolic reactions of thermophilic and hyperthermophilic Archaea and Bacteria. FEMS Microbiology Reviews , 25, 175-243. Aragno, M. (1978). Enrichment, isolation and preliminary characterization of a thermophilic, endospore-forming hydrogen bacterium. FEMS Micobiol. Lett. , 3: 13-15. Aragno, M. (1992). The Thermophilic, Aerobic, Hydrogen-Oxidizing (Knallgas) Bacteria. In A. Balows, H. Trüper, M. Dworkin, W. Harder, & K. Schleifer, The Prokaryotes, a handbook on biology of bacteria. 2nd ed. vol. 4 (pp. 3917-3933.). New York: Springer Verlag. Aragno, M., & Schlegel, H. G. (1992). The mesophilic Hydrogen-Oxidizing (Knallgas) Bacteria. In A. Balows, H. Truper, M. Dworkin, W. Harder, & K.-H. Schleifer, The Prokaryotes 2nd. ed. (pp. 344-384). New York: Springer. Ármannson, H. (2002, May 30-31). Erindi á ráðstefnu um málefni veitufyrirtækja . Grænt bókhald í jarðhita- samanburður á útblæstri við aðra orkugjafa . Akureyri, Iceland: Samorka. Bae, S., Kwak, K., Kim, S., Chung, S., & Igarashi, Y. (2001). Isolation and Characterization of CO2-Fixing Hydrogen -Oxidizing Marine 109 University of Akureyri Department of Natural Resource Science Bacteria. -

Characterization of the Uncommon Enzymes from (2004)
OPEN Mannosylglucosylglycerate biosynthesis SUBJECT AREAS: in the deep-branching phylum WATER MICROBIOLOGY MARINE MICROBIOLOGY Planctomycetes: characterization of the HOMEOSTASIS MULTIENZYME COMPLEXES uncommon enzymes from Rhodopirellula Received baltica 13 March 2013 Sofia Cunha1, Ana Filipa d’Avo´1, Ana Mingote2, Pedro Lamosa3, Milton S. da Costa1,4 & Joana Costa1,4 Accepted 23 July 2013 1Center for Neuroscience and Cell Biology, University of Coimbra, 3004-517 Coimbra, Portugal, 2Instituto de Tecnologia Quı´mica Published e Biolo´gica, Universidade Nova de Lisboa, 2780-157 Oeiras, Portugal, 3Centro de Ressonaˆncia Magne´tica Anto´nio Xavier, 7 August 2013 Instituto de Tecnologia Quı´mica e Biolo´gica, Universidade Nova de Lisboa, 2781-901 Oeiras, Portugal, 4Department of Life Sciences, University of Coimbra, Apartado 3046, 3001-401 Coimbra, Portugal. Correspondence and The biosynthetic pathway for the rare compatible solute mannosylglucosylglycerate (MGG) accumulated by requests for materials Rhodopirellula baltica, a marine member of the phylum Planctomycetes, has been elucidated. Like one of the should be addressed to pathways used in the thermophilic bacterium Petrotoga mobilis, it has genes coding for J.C. ([email protected].) glucosyl-3-phosphoglycerate synthase (GpgS) and mannosylglucosyl-3-phosphoglycerate (MGPG) synthase (MggA). However, unlike Ptg. mobilis, the mesophilic R. baltica uses a novel and very specific MGPG phosphatase (MggB). It also lacks a key enzyme of the alternative pathway in Ptg. mobilis – the mannosylglucosylglycerate synthase (MggS) that catalyses the condensation of glucosylglycerate with GDP-mannose to produce MGG. The R. baltica enzymes GpgS, MggA, and MggB were expressed in E. coli and characterized in terms of kinetic parameters, substrate specificity, temperature and pH dependence. -

And Thermo-Adaptation in Hyperthermophilic Archaea: Identification of Compatible Solutes, Accumulation Profiles, and Biosynthetic Routes in Archaeoglobus Spp
Universidade Nova de Lisboa Osmo- andInstituto thermo de Tecnologia-adaptation Química e Biológica in hyperthermophilic Archaea: Subtitle Subtitle Luís Pedro Gafeira Gonçalves Osmo- and thermo-adaptation in hyperthermophilic Archaea: identification of compatible solutes, accumulation profiles, and biosynthetic routes in Archaeoglobus spp. OH OH OH CDP c c c - CMP O O - PPi O3P P CTP O O O OH OH OH OH OH OH O- C C C O P O O P i Dissertation presented to obtain the Ph.D degree in BiochemistryO O- Instituto de Tecnologia Química e Biológica | Universidade Nova de LisboaP OH O O OH OH OH Oeiras, Luís Pedro Gafeira Gonçalves January, 2008 2008 Universidade Nova de Lisboa Instituto de Tecnologia Química e Biológica Osmo- and thermo-adaptation in hyperthermophilic Archaea: identification of compatible solutes, accumulation profiles, and biosynthetic routes in Archaeoglobus spp. This dissertation was presented to obtain a Ph. D. degree in Biochemistry at the Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa. By Luís Pedro Gafeira Gonçalves Supervised by Prof. Dr. Helena Santos Oeiras, January, 2008 Apoio financeiro da Fundação para a Ciência e Tecnologia (POCI 2010 – Formação Avançada para a Ciência – Medida IV.3) e FSE no âmbito do Quadro Comunitário de apoio, Bolsa de Doutoramento com a referência SFRH / BD / 5076 / 2001. ii ACKNOWNLEDGMENTS The work presented in this thesis, would not have been possible without the help, in terms of time and knowledge, of many people, to whom I am extremely grateful. Firstly and mostly, I need to thank my supervisor, Prof. Helena Santos, for her way of thinking science, her knowledge, her rigorous criticism, and her commitment to science. -

Alvinella Pompejana Is an Endemic Inhabitant Tof Deep-Sea Hydrothermal Vents Located from 21°N to 32°S Latitude on the East Pacific Rise (1)
Metagenome analysis of an extreme microbial symbiosis reveals eurythermal adaptation and metabolic flexibility Joseph J. Grzymskia,1, Alison E. Murraya,1, Barbara J. Campbellb, Mihailo Kaplarevicc, Guang R. Gaoc,d, Charles Leee, Roy Daniele, Amir Ghadirif, Robert A. Feldmanf, and Stephen C. Caryb,d,2 aDivision of Earth and Ecosystem Sciences, Desert Research Institute, 2215 Raggio Parkway, Reno, NV 89512; bCollege of Marine and Earth Studies, University of Delaware, Lewes, DE 19958; cDelaware Biotechnology Institute, 15 Innovation Way, Newark, DE 19702; dElectrical and Computer Engineering, University of Delaware, 140 Evans Hall, Newark, DE 19716; eDepartment of Biological Sciences, University of Waikato, Hamilton, New Zealand; fSymBio Corporation, 1455 Adams Drive, Menlo Park, CA 94025 Edited by George N. Somero, Stanford University, Pacific Grove, CA, and approved September 17, 2008 (received for review March 20, 2008) Hydrothermal vent ecosystems support diverse life forms, many of the thermal tolerance of a structural protein biomarker (5) which rely on symbiotic associations to perform functions integral supports the assertion that A. pompejana is likely among the to survival in these extreme physicochemical environments. Epsi- most thermotolerant and eurythermal metazoans on Earth lonproteobacteria, found free-living and in intimate associations (6, 7). with vent invertebrates, are the predominant vent-associated A. pompejana is characterized by a filamentous microflora that microorganisms. The vent-associated polychaete worm, Alvinella forms cohesive hair-like projections from mucous glands lining pompejana, is host to a visibly dense fleece of episymbionts on its the polychaete’s dorsal intersegmentary spaces (8). The episym- dorsal surface. The episymbionts are a multispecies consortium of biont community is constrained to the bacterial subdivision, Epsilonproteobacteria present as a biofilm. -

Pan-Genome Analyses Identify Lineage
University of Rhode Island DigitalCommons@URI Cell and Molecular Biology Faculty Publications Cell and Molecular Biology 2014 Pan-Genome Analyses Identify Lineage- and Niche-Specific aM rkers of Evolution and Adaptation in Epsilonproteobacteria Ying Zhang University of Rhode Island, [email protected] Stefan M. Sievert Creative Commons License Creative Commons License This work is licensed under a Creative Commons Attribution 3.0 License. Follow this and additional works at: https://digitalcommons.uri.edu/cmb_facpubs Citation/Publisher Attribution Zhang Y., Sievert S.M. (2014). "Pan-genome analyses identify lineage- and niche-specific am rkers of evolution and adaptation in Epsilonproteobacteria." Frontiers in Microbiology. 5: 110. Available at: http://dx.doi.org/10.3389/fmicb.2014.00110 This Article is brought to you for free and open access by the Cell and Molecular Biology at DigitalCommons@URI. It has been accepted for inclusion in Cell and Molecular Biology Faculty Publications by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. ORIGINAL RESEARCH ARTICLE published: 19 March 2014 MICROBIOLOGY doi: 10.3389/fmicb.2014.00110 Pan-genome analyses identify lineage- and niche-specific markers of evolution and adaptation in Epsilonproteobacteria Ying Zhang*† and Stefan M. Sievert Biology Department, Woods Hole Oceanographic Institution, Woods Hole, MA, USA Edited by: The rapidly increasing availability of complete bacterial genomes has created new Martin G. Klotz, University of North opportunities for reconstructing bacterial evolution, but it has also highlighted the difficulty Carolina at Charlotte, USA to fully understand the genomic and functional variations occurring among different Reviewed by: lineages. Using the class Epsilonproteobacteria as a case study, we investigated the Barbara J. -

Lewis M. Ward *, Airi Idei , Mayuko Nakagawa , Yuichiro
bioRxiv preprint doi: https://doi.org/10.1101/428698; this version posted January 27, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Geochemical and metagenomic characterization of Jinata Onsen, a Proterozoic-analog hot spring, 2 reveals novel microbial diversity including iron-tolerant phototrophs and thermophilic lithotrophs 3 4 Lewis M. Ward1,2,3*, Airi Idei4, Mayuko Nakagawa2,5, Yuichiro Ueno2,5,6, Woodward W. 5 Fischer3, Shawn E. McGlynn2* 6 7 1. Department of Earth and Planetary Sciences, Harvard University, Cambridge, MA 02138 USA 8 2. Earth-Life Science Institute, Tokyo Institute of Technology, Meguro, Tokyo, 152-8550, Japan 9 3. Division of Geological and Planetary Sciences, California Institute of Technology, Pasadena, CA 10 91125 USA 11 4. Department of Biological Sciences, Tokyo Metropolitan University, Hachioji, Tokyo 192-0397, 12 Japan 13 5. Department of Earth and Planetary Sciences, Tokyo Institute of Technology, Meguro, Tokyo, 14 152-8551, Japan 15 6. Department of Subsurface Geobiological Analysis and Research, Japan Agency for Marine-Earth 16 Science and Technology, Natsushima-cho, Yokosuka 237-0061, Japan 17 Correspondence: [email protected] or [email protected] 18 19 Abstract 20 Hydrothermal systems, including terrestrial hot springs, contain diverse geochemical 21 conditions that vary over short spatial scales due to progressive interaction between the reducing 22 hydrothermal fluids, the oxygenated atmosphere, and in some cases seawater. At Jinata Onsen, 23 on Shikinejima Island, Japan, an intertidal, anoxic, iron-rich hot spring mixes with the 24 oxygenated atmosphere and seawater over short spatial scales, creating a diversity of chemical 25 potentials and redox pairs over a distance ~10 m. -

Hydrogen Oxidation Coupled to Sulfur Reduction in Deep-Sea Vent Bacteria. Francesco Smedile1, Ashley Grosche1, Sushmita Patwardh
Hydrogen oxidation coupled to sulfur reduction in deep-sea vent bacteria. Francesco Smedile1, Ashley Grosche1, Sushmita Patwardhan1, Donato Giovannelli1,2, Ben Jelen1, Costantino Vetriani1 1Department of Biochemistry and Microbiology and Department of Marine and Coastal Sciences, Rutgers University, New Brunswick, NJ 08901, USA; [email protected] 2Earth-Life Science Institute, Tokyo Institute of Technology, Tokyo 152-8551, Japan Among deep-sea ecosystems, hydrothermal vents represent “relic” environments that resemble the early Earth. The genomes of modern vent microorganisms carry both ancestral and recently acquired genes providing excellent models to reconstruct how microbial metabolism co-evolved with our planet. Nautiliaceae (class Epsilonproteobacteria) and Desulfurobacteraceae (class Aquificae) are two key groups of bacteria in marine geothermal habitats. Although these bacteria are phylogenetically distant and colonize different temperature niches, they share key central metabolic characteristics: both fix CO2 and obtain energy coupling the oxidation of H2 to the reduction of elemental sulfur and/or nitrate. While nitrate reduction has been investigated, the sulfur reduction pathways have yet to be fully resolved. Focusing on the genomes of members of the Nautiliaceae, Cetia pacifica, Nautilia profundicola Caminibacter mediatlanticus, as well as the genomes of members of the Desulfurobacteraceae, Thermovibrio ammonificans strains HB-1 and HB-5, Desulfurobacterium thermolithotrophum and Phorcysia thermohydrogeniphila, we reported the distribution and phylogenetic reconstruction of four enzymes possibly involved in sulfur reduction. The genes encoding for polysulfide reductase and sulfide quinone oxidoreductase were identified in both the Nautiliaceae and the Desulfurobacteraceae, while the sulfhydrogenase II and the NAD/FAD-dependent reductase were identified in the Desulfurobacteraceae. Comparative genomic analyses were used to formulate hypotheses on how H2 oxidation can be coupled with sulfur reduction. -

Metagenomic Insights Into S(0) Precipitation in a Terrestrial Subsurface Lithoautotrophic Ecosystem
ORIGINAL RESEARCH ARTICLE published: 08 January 2015 doi: 10.3389/fmicb.2014.00756 Metagenomic insights into S(0) precipitation in a terrestrial subsurface lithoautotrophic ecosystem Trinity L. Hamilton 1*, Daniel S. Jones 1,2, Irene Schaperdoth 1 and Jennifer L. Macalady 1* 1 Department of Geosciences, Penn State Astrobiology Research Center, The Pennsylvania State University, University Park, PA, USA 2 Department of Earth Sciences, University of Minnesota, Minneapolis, MN, USA Edited by: The Frasassi and Acquasanta Terme cave systems in Italy host isolated lithoautotrophic John R. Spear, Colorado School of ecosystems characterized by sulfur-oxidizing biofilms with up to 50% S(0) by mass. Mines, USA The net contributions of microbial taxa in the biofilms to production and consumption Reviewed by: of S(0) are poorly understood and have implications for understanding the formation Matthew Schrenk, Michigan State University, USA of geological sulfur deposits as well as the ecological niches of sulfur-oxidizing John R. Spear, Colorado School of autotrophs. Filamentous Epsilonproteobacteria are among the principal biofilm architects Mines, USA in Frasassi and Acquasanta Terme streams, colonizing high-sulfide, low-oxygen niches Takuro Nunoura, Japan Agency for relative to other major biofilm-forming populations. Metagenomic sequencing of eight Marine-Earth Science & Technology, Japan biofilm samples indicated the presence of diverse and abundant Epsilonproteobacteria. *Correspondence: Populations of Sulfurovum-like organisms were the most abundant Epsilonproteobacteria Trinity L. Hamilton and Jennifer L. regardless of differences in biofilm morphology, temperature, or water chemistry. After Macalady, Department of assembling and binning the metagenomic data, we retrieved four nearly-complete Geosciences, Penn State genomes of Sulfurovum-like organisms as well as a Sulfuricurvum spp. -
Endomicrobia
Environment GCF 001642875 4 16S 1 Mariniblastus fucicola GCF 001642875 GCF 002277955 1 16S 1 Thermogutta terrifontis GCF 002277955 GCF 000255655 9 16S 1 Schlesneria paludicola DSM 18645 GCF 000255655 Freshwater GCF 001610835 1 16S 2 Planctomyces sp SH PL14 GCF 001610835 GCF 001610835 1 16S 1 Planctomyces sp SH PL14 GCF 001610835 GCF 000165715 1 16S 1 Rubinisphaera brasiliensis DSM 5305 GCF 000165715 Tree scale: 0.1 GCF 000165715 1 16S 2 Rubinisphaera brasiliensis DSM 5305 GCF 000165715 GCF 900129635 4 16S 3 Singulisphaera sp GP187 GCF 900129635 Soil GCF 900129635 4 16S 2 Singulisphaera sp GP187 GCF 900129635 GCF 900129635 4 16S 1 Singulisphaera sp GP187 GCF 900129635 GCF 900129635 5 16S 1 Singulisphaera sp GP187 GCF 900129635 GCF 900129635 5 16S 3 Singulisphaera sp GP187 GCF 900129635 Sediment GCF 900129635 5 16S 4 Singulisphaera sp GP187 GCF 900129635 GCF 900129635 5 16S 5 Singulisphaera sp GP187 GCF 900129635 GCF 900129635 5 16S 2 Singulisphaera sp GP187 GCF 900129635 GCF 000417735 1 16S 1 Chlamydia psittaci 10 881 SC42 GCF 000417735 Gut GCF 000750955 17 16S 1 Criblamydia sequanensis CRIB 18 GCF 000750955 GCF 900000175 28 16S 1 Estrella lausannensis GCF 900000175 GCF 000092785 1 16S 2 Waddlia chondrophila WSU 86 1044 GCF 000092785 GCF 000092785 1 16S 1 Waddlia chondrophila WSU 86 1044 GCF 000092785 GCF 000237205 1 16S 1 Simkania negevensis Z GCF 000237205 Oil GCA 000756735 1 16S 2 Candidatus Rubidus massiliensis GCA 000756735 GCA 000756735 1 16S 1 Candidatus Rubidus massiliensis GCA 000756735 GCF 001545115 16 16S 1 Parachlamydia sp