Predicting Virulence Factors in Filamentous Fungi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genomes of Polyextremophilic Cyanidiales Contain 1% 2 Horizontally Transferred Genes with Diverse Adaptive Functions 3 4 Alessandro W
bioRxiv preprint doi: https://doi.org/10.1101/526111; this version posted January 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 The genomes of polyextremophilic Cyanidiales contain 1% 2 horizontally transferred genes with diverse adaptive functions 3 4 Alessandro W. Rossoni1#, Dana C. Price2, Mark Seger3, Dagmar Lyska1, Peter Lammers3, 5 Debashish Bhattacharya4 & Andreas P.M. Weber1* 6 7 1Institute of Plant Biochemistry, Cluster of Excellence on Plant Sciences (CEPLAS), Heinrich 8 Heine University, Universitätsstraße 1, 40225 Düsseldorf, Germany 9 2Department of Plant Biology, Rutgers University, New Brunswick, NJ 08901, USA 10 3Arizona Center for Algae Technology and Innovation, Arizona State University, Mesa, AZ 11 85212, USA 12 4Department of Biochemistry and Microbiology, Rutgers University, New Brunswick, NJ 13 08901, USA 14 15 *Corresponding author: Prof. Dr. Andreas P.M. Weber, 16 e-mail: [email protected] 17 bioRxiv preprint doi: https://doi.org/10.1101/526111; this version posted January 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 18 Abstract 19 The role and extent of horizontal gene transfer (HGT) in eukaryotes are hotly disputed topics 20 that impact our understanding regarding the origin of metabolic processes and the role of 21 organelles in cellular evolution. -
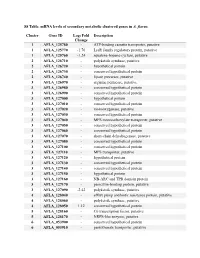
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

Comparative Genomics of Bradyrhizobium Japonicum CPAC
Siqueira et al. BMC Genomics 2014, 15:420 http://www.biomedcentral.com/1471-2164/15/420 RESEARCH ARTICLE Open Access Comparative genomics of Bradyrhizobium japonicum CPAC 15 and Bradyrhizobium diazoefficiens CPAC 7: elite model strains for understanding symbiotic performance with soybean Arthur Fernandes Siqueira1,2†, Ernesto Ormeño-Orrillo3†,RangelCelsoSouza4,ElisetePainsRodrigues5, Luiz Gonzaga Paula Almeida4, Fernando Gomes Barcellos5, Jesiane Stefânia Silva Batista6, Andre Shigueyoshi Nakatani2, Esperanza Martínez-Romero3, Ana Tereza Ribeiro Vasconcelos4 and Mariangela Hungria1,2* Abstract Background: The soybean-Bradyrhizobium symbiosis can be highly efficient in fixing nitrogen, but few genomic sequences of elite inoculant strains are available. Here we contribute with information on the genomes of two commercial strains that are broadly applied to soybean crops in the tropics. B. japonicum CPAC 15 (=SEMIA 5079) is outstanding in its saprophytic capacity and competitiveness, whereas B. diazoefficiens CPAC 7 (=SEMIA 5080) is known for its high efficiency in fixing nitrogen. Both are well adapted to tropical soils. The genomes of CPAC 15 and CPAC 7 were compared to each other and also to those of B. japonicum USDA 6T and B. diazoefficiens USDA 110T. Results: Differences in genome size were found between species, with B. japonicum having larger genomes than B. diazoefficiens. Although most of the four genomes were syntenic, genome rearrangements within and between species were observed, including events in the symbiosis island. In addition to the symbiotic region, several genomic islands were identified. Altogether, these features must confer high genomic plasticity that might explain adaptation and differences in symbiotic performance. It was not possible to attribute known functions to half of the predicted genes. -

Purification and Characterization of a Novel Nitrilase of Rhodococcus
JOURNAL OF BACTERIOLOGY, Sept. 1990, p. 4807-4815 Vol. 172, No. 9 0021-9193/90/094807-09$02.00/0 Copyright X3 1990, American Society for Microbiology Purification and Characterization of a Novel Nitrilase of Rhodococcus rhodochrous K22 That Acts on Aliphatic Nitriles MICHIHIKO KOBAYASHI,* NORIYUKI YANAKA, TORU NAGASAWA, AND HIDEAKI YAMADA Department ofAgricultural Chemistry, Faculty ofAgriculture, Kyoto University, Sakyo-ku, Kyoto 606, Japan Received 18 April 1990/Accepted 8 June 1990 A novel nitrilase that preferentially catalyzes the hydrolysis of aliphatic nitriles to the corresponding carboxylic acids and ammonia was found in the cells of a facultative crotononitrile-utilizing actinomycete isolated from soil. The strain was taxonomically studied and identified as Rhodococcus rhodochrous. The nitrilase was purified, with 9.08% overall recovery, through five steps from a cell extract of the stain. After the last step, the purified enzyme appeared to be homogeneous, as judged by polyacrylamide gel electrophoresis, analytical centrifugation, and double immunodiffusion in agarose. The relative molecular weight values for the native enzyme, estimated from the ultracentrifugal equilibrium and by high-performance liquid chromatog- raphy, were approximately 604,000 + 30,000 and 650,000, respectively, and the enzyme consisted of 15 to 16 subunits identical in molecular weight (41,000). The enzyme acted on aliphatic olefinic nitriles such as crotononitrile and acrylonitrile as the most suitable substrates. The apparent Km values for crotononitrile and acrylonitrile were 18.9 and 1.14 mM, respectively. The nitrilase also catalyzed the direct hydrolysis of saturated aliphatic nitriles, such as valeronitrile, 4-chlorobutyronitrile, and glutaronitrile, to the correspond- ing acids without the formation of amide intermediates. -

Direct Link to Fulltext
ISOLATION, IDENTIFICATION AND SUBSTRATE SPECIFICITY OF A NITRILASE PRODUCING BACTERIA, Acidovorax sp. SK1 Soumya Koippully, Subha Swaraj Pattnaik, Siddhardha Busi* Address(es): Department of Microbiology, School of Life Sciences, Pondicherry University, Puducherry-605 014, India. *Corresponding author: [email protected] doi: 10.15414/jmbfs.2018.8.2.788-793 ARTICLE INFO ABSTRACT Received 13. 5. 2018 The process of biocatalysis or biotransformation remains the core of industrial biotechnology owing to its importance in the synthesis of Revised 3. 9. 2018 high-value products in a cost effective manner. Nitrile biotransformation has also achieved considerable attention in last few decades Accepted 5. 9. 2018 due to its widespread biotechnological and industrial applications. In the present study, a versatile, potent nitrile-degrading bacterium, Published 1. 10. 2018 Acidovorax sp. SK1 was isolated from the cracker waste dumping site of Sivakasi, Tamilnadu and characterized for its biocatalytic potential, nitrilase production and enzyme activity. A pH sensitive indicator-based assay was performed to identify the nitrile degrading ability of the isolated samples. Semi quantitative High performance thin layer chromatographic (HPTLC) method was performed for Regular article quantitative measurement of mandelic acid produced from degradation of nitrile compound, mandelonitrile. The optimization of medium and nutritional parameters were studied for the improvement of nitrilase activity, which indicated that maximum nitrilase activity was observed at an optimum pH of 7.0, agitation at 100 rpm, glucose as best carbon source (10 g/L) and yeast extract (0.1 g/L) as principal nitrogen source. Biomass is also a critical parameter in the biocatalysis of mandelonitrile to mandelic acid and at a biomass of 100 mg/L, maximum nitrilase activity of 0.026 I.U was observed. -

Supplementary Material Supplementary Results And
R. D. Barabote SUPPLEMENTARY MATERIAL SUPPLEMENTARY RESULTS AND DISCUSSION tRNA and codon usage. Forty-five tRNAs representing 43 different anticodons are encoded in the genome (Supplementary Table S1). The tRNAMet is present in three copies in the genome. In contrast to the number of tRNAs, all 61 sense codons are encoded in the genome sequence. The codon usage correlates well with the tRNA complement and is consistent with the high G+C content of the genome as the GC-rich codons predominate in the organism (Supplementary Table S1). Codons ATA (Ile), CGC (Arg), and CGA (Arg) as well as all codons that have a T at the third position, with the exception of CGT (Arg), do not appear to have a cognate tRNA in A. cellulolyticus. As a likely evolutionary adaptation to the available tRNAs for any given amino acid, codons that do not have a cognate tRNA occur with the least frequency in the A. cellulolyticus genome when compared to synonymous codons differing just in the 3rd position. However, the exceptions to these are for glycine (GGT (18.8%) > GGA (14.4%)), leucine (CTT (10.6%) > CTA (1.4%)), arginine (CGC (33.3%) > CGT (11.1%)), and valine (GTT (10.4%) > GTA (3.8%)). The relative preference for CGC codon over CGT codon follows the high G+C content of the genome, while the remaining four biases mentioned above may simply reflect evolutionary conservation of codon usage, as a similar trend is seen in Frankia (Supplementary Table S2). The functional significance of this bias remains elusive. The A. cellulolyticus genome encodes a parsimonious complement of 46 tRNAs. -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000725805Cyc: Streptomyces xanthophaeus Cellular Overview Connections between pathways are omitted for legibility. -

Genetic Characterisaton of Rhodococcus Rhodochrous ATCC
The copyright of this thesis vests in the author. No quotation from it or information derived from it is to be published without full acknowledgement of the source. The thesis is to be used for private study or non- commercial research purposes only. Published by the University of Cape Town (UCT) in terms of the non-exclusive license granted to UCT by the author. University of Cape Town Genetic characterization of Rhodococcus rhodochrous ATCC BAA-870 with emphasis on nitrile hydrolysing enzymes n ow Joni Frederick A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the Departmentty of of MolecularCape and T Cell Biology, Universitysi of Cape Town er UnivSupervisor: Professor B. T. Sewell Co-supervisor: Professor D. Brady February 2013 Keywords Nitrile hydrolysis Biocatalysis Rhodococcus rhodochrous ATCC BAA-870 Genome sequencing Nitrilase Nitrile hydratase n ow ty of Cape T si er Univ ii Keywords Abstract Rhodococcus rhodochrous ATCC BAA-870 (BAA-870) had previously been isolated on selective media for enrichment of nitrile hydrolysing bacteria. The organism was found to have a wide substrate range, with activity against aliphatics, aromatics, and aryl aliphatics, and enantioselectivity towards beta substituted nitriles and beta amino nitriles, compounds that have potential applications in the pharmaceutical industry. This makes R. rhodochrous ATCC BAA-870 potentially a versatile biocatalyst for the synthesis of a broad range of compounds with amide and carboxylic acid groups that can be derived from structurally related nitrile precursors. The selectivity of biocatalysts allows for high product yields and better atom economyn than non- selective chemical methods of performing this reaction, suchow as acid or base hydrolysis. -

Comparison of Nitrile Hydratases in Rhodococcus Rhodochrous DAP 96253 and DAP 96622 Growing on Inducing and Non- Inducing Media
Georgia State University ScholarWorks @ Georgia State University Biology Dissertations Department of Biology Spring 4-26-2013 Comparison of Nitrile Hydratases in Rhodococcus Rhodochrous DAP 96253 and DAP 96622 Growing on Inducing and Non- Inducing Media Fengkun Du Georgia State University Follow this and additional works at: https://scholarworks.gsu.edu/biology_diss Recommended Citation Du, Fengkun, "Comparison of Nitrile Hydratases in Rhodococcus Rhodochrous DAP 96253 and DAP 96622 Growing on Inducing and Non-Inducing Media." Dissertation, Georgia State University, 2013. https://scholarworks.gsu.edu/biology_diss/130 This Dissertation is brought to you for free and open access by the Department of Biology at ScholarWorks @ Georgia State University. It has been accepted for inclusion in Biology Dissertations by an authorized administrator of ScholarWorks @ Georgia State University. For more information, please contact [email protected]. COMPARISON OF NITRILE HYDRATASES IN RHODOCOCCUS RHODOCHROUS DAP 96253 AND DAP 96622 GROWING ON INDUCING AND NON INDUCING MEDIA by FENGKUN DU Under the Direction of George E. Pierce ABSTRACT Nitrile hydratase activity in Rhodococcus rhodochrous DAP 96253 can be induced with multiple inducers that include urea, cobalt (Co), iron (Fe) and nickel (Ni). When induced with Co/urea, cells of R. rhodochrous DAP 96253 expressed the highest level of nitrile hydratase activity (~200 units/min·mg-cdw) when compared with the other inducers tested. Cells induced with Co had the second highest nitrile hydratase activity (~7 units/min·mg-cdw), whereas in the uninduced cells, nitrile hydratase activity was lower than 1 unit/min·mg-cdw. Similarly in R. rhodochrous DAP 96622, when induced with Co/urea, the nitrile hydratase activity of R. -

Analysis of the Sll0783 Function in PHB Synthesis in Synechocystis PCC 6803: a Crucial Role of NADPH
Analysis of the Sll0783 Function in PHB Synthesis in Synechocystis PCC 6803: a Crucial Role of NADPH in N-Starvation Dissertation der Mathematisch-Naturwissenschaftlichen Fakultät der Eberhard Karls Universität Tübingen zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) vorgelegt von Maximilian Schlebusch aus Engelskirchen Tübingen 2012 Tag der mündlichen Qualifikation: 03.02.2012 Dekan: Prof. Dr. Wolfgang Rosenstiel 1. Berichterstatter: Prof. Dr. Karl Forchhammer 2. Berichterstatter: Prof. Dr. Wolfgang Wohlleben Abstract Nitrogen frequently is a limiting nutrient in natural habitats. Therefore, cyanobac- teria as well as other autotrophic organisms have developed multiple strategies to adapt to nitrogen deficiency. Transcriptomic analyses of the strain Synechocys- tis PCC 6803 under nitrogen-deficient conditions revealed a highly induced gene (sll0783 ), which is annotated as conserved protein with unknown function. This gene is part of a cluster with seven genes and in the upstream region lies a pre- dicted NtcA-binding site. Homologues of this cluster occur in some unicellular, non-diazotrophic cyanobacteria, in several α-, β- and γ-proteobacteria as well as in some gram-positives. The common link between the heterotrophic bacteria seems to be the ability of nitrogen fixation and production of polyhydroxybu- tyrate (PHB), whereas among the cyanobacteria only Synechocystis PCC 6803 can accumulate PHB. In this work, a knockout mutant of this gene in Synechocystis PCC 6803 was characterised. This mutant was unable to accumulate PHB, a carbon and en- ergy storage compound. The levels of precursor metabolites such as glycogen and acetyl-CoA were not reduced. The impairment in PHB accumulation cor- related with a loss of PHB synthase activity during prolonged nitrogen starva- tion. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

The Complete Genome Sequence of the Nitrile
Frederick et al. BMC Genomics (2020) 21:3 https://doi.org/10.1186/s12864-019-6405-7 RESEARCH ARTICLE Open Access The complete genome sequence of the nitrile biocatalyst Rhodococcus rhodochrous ATCC BAA-870 Joni Frederick1,2,3, Fritha Hennessy1, Uli Horn4, Pilar de la Torre Cortés5, Marcel van den Broek5, Ulrich Strych6,7, Richard Willson6,8, Charles A. Hefer9,10, Jean-Marc G. Daran5, Trevor Sewell2, Linda G. Otten11* and Dean Brady1,12* Abstract Background: Rhodococci are industrially important soil-dwelling Gram-positive bacteria that are well known for both nitrile hydrolysis and oxidative metabolism of aromatics. Rhodococcus rhodochrous ATCC BAA-870 is capable of metabolising a wide range of aliphatic and aromatic nitriles and amides. The genome of the organism was sequenced and analysed in order to better understand this whole cell biocatalyst. Results: The genome of R. rhodochrous ATCC BAA-870 is the first Rhodococcus genome fully sequenced using Nanopore sequencing. The circular genome contains 5.9 megabase pairs (Mbp) and includes a 0.53 Mbp linear plasmid, that together encode 7548 predicted protein sequences according to BASys annotation, and 5535 predicted protein sequences according to RAST annotation. The genome contains numerous oxidoreductases, 15 identified antibiotic and secondary metabolite gene clusters, several terpene and nonribosomal peptide synthetase clusters, as well as 6 putative clusters of unknown type. The 0.53 Mbp plasmid encodes 677 predicted genes and contains the nitrile converting gene cluster, including a nitrilase, a low molecular weight nitrile hydratase, and an enantioselective amidase. Although there are fewer biotechnologically relevant enzymes compared to those found in rhodococci with larger genomes, such as the well-known Rhodococcus jostii RHA1, the abundance of transporters in combination with the myriad of enzymes found in strain BAA-870 might make it more suitable for use in industrially relevant processes than other rhodococci.