The Genomes of Polyextremophilic Cyanidiales Contain 1% 2 Horizontally Transferred Genes with Diverse Adaptive Functions 3 4 Alessandro W
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METABOLIC EVOLUTION in GALDIERIA SULPHURARIA By
METABOLIC EVOLUTION IN GALDIERIA SULPHURARIA By CHAD M. TERNES Bachelor of Science in Botany Oklahoma State University Stillwater, Oklahoma 2009 Submitted to the Faculty of the Graduate College of the Oklahoma State University in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY May, 2015 METABOLIC EVOLUTION IN GALDIERIA SUPHURARIA Dissertation Approved: Dr. Gerald Schoenknecht Dissertation Adviser Dr. David Meinke Dr. Andrew Doust Dr. Patricia Canaan ii Name: CHAD M. TERNES Date of Degree: MAY, 2015 Title of Study: METABOLIC EVOLUTION IN GALDIERIA SULPHURARIA Major Field: PLANT SCIENCE Abstract: The thermoacidophilic, unicellular, red alga Galdieria sulphuraria possesses characteristics, including salt and heavy metal tolerance, unsurpassed by any other alga. Like most plastid bearing eukaryotes, G. sulphuraria can grow photoautotrophically. Additionally, it can also grow solely as a heterotroph, which results in the cessation of photosynthetic pigment biosynthesis. The ability to grow heterotrophically is likely correlated with G. sulphuraria ’s broad capacity for carbon metabolism, which rivals that of fungi. Annotation of the metabolic pathways encoded by the genome of G. sulphuraria revealed several pathways that are uncharacteristic for plants and algae, even red algae. Phylogenetic analyses of the enzymes underlying the metabolic pathways suggest multiple instances of horizontal gene transfer, in addition to endosymbiotic gene transfer and conservation through ancestry. Although some metabolic pathways as a whole appear to be retained through ancestry, genes encoding individual enzymes within a pathway were substituted by genes that were acquired horizontally from other domains of life. Thus, metabolic pathways in G. sulphuraria appear to be composed of a ‘metabolic patchwork’, underscored by a mosaic of genes resulting from multiple evolutionary processes. -

The Genome of Prasinoderma Coloniale Unveils the Existence of a Third Phylum Within Green Plants
SUPPLEMENTARY INFORMATIONARTICLES https://doi.org/10.1038/s41559-020-1221-7 In the format provided by the authors and unedited. The genome of Prasinoderma coloniale unveils the existence of a third phylum within green plants Linzhou Li1,2,13, Sibo Wang1,3,13, Hongli Wang1,4, Sunil Kumar Sahu 1, Birger Marin 5, Haoyuan Li1, Yan Xu1,4, Hongping Liang1,4, Zhen Li 6, Shifeng Cheng1, Tanja Reder5, Zehra Çebi5, Sebastian Wittek5, Morten Petersen3, Barbara Melkonian5,7, Hongli Du8, Huanming Yang1, Jian Wang1, Gane Ka-Shu Wong 1,9, Xun Xu 1,10, Xin Liu 1, Yves Van de Peer 6,11,12 ✉ , Michael Melkonian5,7 ✉ and Huan Liu 1,3 ✉ 1State Key Laboratory of Agricultural Genomics, BGI-Shenzhen, Shenzhen, China. 2Department of Biotechnology and Biomedicine, Technical University of Denmark, Lyngby, Denmark. 3Department of Biology, University of Copenhagen, Copenhagen, Denmark. 4BGI Education Center, University of Chinese Academy of Sciences, Shenzhen, China. 5Institute for Plant Sciences, Department of Biological Sciences, University of Cologne, Cologne, Germany. 6Department of Plant Biotechnology and Bioinformatics (Ghent University) and Center for Plant Systems Biology, Ghent, Belgium. 7Central Collection of Algal Cultures, Faculty of Biology, University of Duisburg-Essen, Essen, Germany. 8School of Biology and Biological Engineering, South China University of Technology, Guangzhou, China. 9Department of Biological Sciences and Department of Medicine, University of Alberta, Edmonton, Alberta, Canada. 10Guangdong Provincial Key Laboratory of Genome Read and Write, BGI-Shenzhen, Shenzhen, China. 11College of Horticulture, Nanjing Agricultural University, Nanjing, China. 12Centre for Microbial Ecology and Genomics, Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria, South Africa. -

Phylogenomics Provides Robust Support for a Two-Domains Tree of Life
ARTICLES https://doi.org/10.1038/s41559-019-1040-x Phylogenomics provides robust support for a two-domains tree of life Tom A. Williams! !1*, Cymon J. Cox! !2, Peter G. Foster3, Gergely J. Szöllősi4,5,6 and T. Martin Embley7* Hypotheses about the origin of eukaryotic cells are classically framed within the context of a universal ‘tree of life’ based on conserved core genes. Vigorous ongoing debate about eukaryote origins is based on assertions that the topology of the tree of life depends on the taxa included and the choice and quality of genomic data analysed. Here we have reanalysed the evidence underpinning those claims and apply more data to the question by using supertree and coalescent methods to interrogate >3,000 gene families in archaea and eukaryotes. We find that eukaryotes consistently originate from within the archaea in a two-domains tree when due consideration is given to the fit between model and data. Our analyses support a close relation- ship between eukaryotes and Asgard archaea and identify the Heimdallarchaeota as the current best candidate for the closest archaeal relatives of the eukaryotic nuclear lineage. urrent hypotheses about eukaryotic origins generally pro- Indeed, it has previously been suggested that it is the 3D tree, rather pose at least two partners in that process: a bacterial endo- than the 2D tree, that is an artefact of long-branch attraction5,9–11, symbiont that became the mitochondrion and a host cell for both because analyses under better-fitting models have recovered C 1–4 that endosymbiosis . The identity of the host has been informed a 2D tree but also because the 3D topology is one in which the two by analyses of conserved genes for the transcription and transla- longest branches in the tree of life—the stems leading to bacteria and tion machinery that are considered essential for cellular life5. -
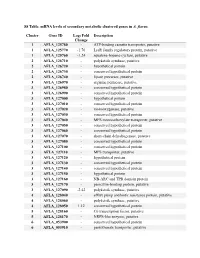
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

Toxicity, Physiological, and Ultrastructural Effects of Arsenic
International Journal of Environmental Research and Public Health Article Toxicity, Physiological, and Ultrastructural Effects of Arsenic and Cadmium on the Extremophilic Microalga Chlamydomonas acidophila Silvia Díaz 1, Patricia De Francisco 1,2, Sanna Olsson 3 , Ángeles Aguilera 2,*, Elena González-Toril 2 and Ana Martín-González 1 1 Department of Genetics, Physiology and Microbiology, Faculty of Biology, Universidad Complutense de Madrid (UCM), C/José Antonio Novais, 12, 28040 Madrid, Spain; [email protected] (S.D.); [email protected] (P.d.F.); [email protected] (A.M.-G.) 2 Astrobiology Center (INTA-CSIC), Carretera de Ajalvir km 4, Torrejón de Ardoz, 28850 Madrid, Spain; [email protected] 3 Department of Forest Ecology and Genetics, INIA Forest Research Center (INIA-CIFOR), Carretera de A Coruña km 7.5, 28040 Madrid, Spain; sanna.olsson@helsinki.fi * Correspondence: [email protected]; Tel.: +34-91-520-6434 Received: 17 December 2019; Accepted: 24 February 2020; Published: 3 March 2020 Abstract: The cytotoxicity of cadmium (Cd), arsenate (As(V)), and arsenite (As(III)) on a strain of Chlamydomonas acidophila, isolated from the Rio Tinto, an acidic environment containing high metal(l)oid concentrations, was analyzed. We used a broad array of methods to produce complementary information: cell viability and reactive oxygen species (ROS) generation measures, ultrastructural observations, transmission electron microscopy energy dispersive x-ray microanalysis (TEM–XEDS), and gene expression. This acidophilic microorganism was affected differently by the tested metal/metalloid: It showed high resistance to arsenic while Cd was the most toxic heavy metal, showing an LC50 = 1.94 µM. -

Comparative Genomics of Bradyrhizobium Japonicum CPAC
Siqueira et al. BMC Genomics 2014, 15:420 http://www.biomedcentral.com/1471-2164/15/420 RESEARCH ARTICLE Open Access Comparative genomics of Bradyrhizobium japonicum CPAC 15 and Bradyrhizobium diazoefficiens CPAC 7: elite model strains for understanding symbiotic performance with soybean Arthur Fernandes Siqueira1,2†, Ernesto Ormeño-Orrillo3†,RangelCelsoSouza4,ElisetePainsRodrigues5, Luiz Gonzaga Paula Almeida4, Fernando Gomes Barcellos5, Jesiane Stefânia Silva Batista6, Andre Shigueyoshi Nakatani2, Esperanza Martínez-Romero3, Ana Tereza Ribeiro Vasconcelos4 and Mariangela Hungria1,2* Abstract Background: The soybean-Bradyrhizobium symbiosis can be highly efficient in fixing nitrogen, but few genomic sequences of elite inoculant strains are available. Here we contribute with information on the genomes of two commercial strains that are broadly applied to soybean crops in the tropics. B. japonicum CPAC 15 (=SEMIA 5079) is outstanding in its saprophytic capacity and competitiveness, whereas B. diazoefficiens CPAC 7 (=SEMIA 5080) is known for its high efficiency in fixing nitrogen. Both are well adapted to tropical soils. The genomes of CPAC 15 and CPAC 7 were compared to each other and also to those of B. japonicum USDA 6T and B. diazoefficiens USDA 110T. Results: Differences in genome size were found between species, with B. japonicum having larger genomes than B. diazoefficiens. Although most of the four genomes were syntenic, genome rearrangements within and between species were observed, including events in the symbiosis island. In addition to the symbiotic region, several genomic islands were identified. Altogether, these features must confer high genomic plasticity that might explain adaptation and differences in symbiotic performance. It was not possible to attribute known functions to half of the predicted genes. -

Purification and Characterization of a Novel Nitrilase of Rhodococcus
JOURNAL OF BACTERIOLOGY, Sept. 1990, p. 4807-4815 Vol. 172, No. 9 0021-9193/90/094807-09$02.00/0 Copyright X3 1990, American Society for Microbiology Purification and Characterization of a Novel Nitrilase of Rhodococcus rhodochrous K22 That Acts on Aliphatic Nitriles MICHIHIKO KOBAYASHI,* NORIYUKI YANAKA, TORU NAGASAWA, AND HIDEAKI YAMADA Department ofAgricultural Chemistry, Faculty ofAgriculture, Kyoto University, Sakyo-ku, Kyoto 606, Japan Received 18 April 1990/Accepted 8 June 1990 A novel nitrilase that preferentially catalyzes the hydrolysis of aliphatic nitriles to the corresponding carboxylic acids and ammonia was found in the cells of a facultative crotononitrile-utilizing actinomycete isolated from soil. The strain was taxonomically studied and identified as Rhodococcus rhodochrous. The nitrilase was purified, with 9.08% overall recovery, through five steps from a cell extract of the stain. After the last step, the purified enzyme appeared to be homogeneous, as judged by polyacrylamide gel electrophoresis, analytical centrifugation, and double immunodiffusion in agarose. The relative molecular weight values for the native enzyme, estimated from the ultracentrifugal equilibrium and by high-performance liquid chromatog- raphy, were approximately 604,000 + 30,000 and 650,000, respectively, and the enzyme consisted of 15 to 16 subunits identical in molecular weight (41,000). The enzyme acted on aliphatic olefinic nitriles such as crotononitrile and acrylonitrile as the most suitable substrates. The apparent Km values for crotononitrile and acrylonitrile were 18.9 and 1.14 mM, respectively. The nitrilase also catalyzed the direct hydrolysis of saturated aliphatic nitriles, such as valeronitrile, 4-chlorobutyronitrile, and glutaronitrile, to the correspond- ing acids without the formation of amide intermediates. -

Direct Link to Fulltext
ISOLATION, IDENTIFICATION AND SUBSTRATE SPECIFICITY OF A NITRILASE PRODUCING BACTERIA, Acidovorax sp. SK1 Soumya Koippully, Subha Swaraj Pattnaik, Siddhardha Busi* Address(es): Department of Microbiology, School of Life Sciences, Pondicherry University, Puducherry-605 014, India. *Corresponding author: [email protected] doi: 10.15414/jmbfs.2018.8.2.788-793 ARTICLE INFO ABSTRACT Received 13. 5. 2018 The process of biocatalysis or biotransformation remains the core of industrial biotechnology owing to its importance in the synthesis of Revised 3. 9. 2018 high-value products in a cost effective manner. Nitrile biotransformation has also achieved considerable attention in last few decades Accepted 5. 9. 2018 due to its widespread biotechnological and industrial applications. In the present study, a versatile, potent nitrile-degrading bacterium, Published 1. 10. 2018 Acidovorax sp. SK1 was isolated from the cracker waste dumping site of Sivakasi, Tamilnadu and characterized for its biocatalytic potential, nitrilase production and enzyme activity. A pH sensitive indicator-based assay was performed to identify the nitrile degrading ability of the isolated samples. Semi quantitative High performance thin layer chromatographic (HPTLC) method was performed for Regular article quantitative measurement of mandelic acid produced from degradation of nitrile compound, mandelonitrile. The optimization of medium and nutritional parameters were studied for the improvement of nitrilase activity, which indicated that maximum nitrilase activity was observed at an optimum pH of 7.0, agitation at 100 rpm, glucose as best carbon source (10 g/L) and yeast extract (0.1 g/L) as principal nitrogen source. Biomass is also a critical parameter in the biocatalysis of mandelonitrile to mandelic acid and at a biomass of 100 mg/L, maximum nitrilase activity of 0.026 I.U was observed. -

Supplementary Material Supplementary Results And
R. D. Barabote SUPPLEMENTARY MATERIAL SUPPLEMENTARY RESULTS AND DISCUSSION tRNA and codon usage. Forty-five tRNAs representing 43 different anticodons are encoded in the genome (Supplementary Table S1). The tRNAMet is present in three copies in the genome. In contrast to the number of tRNAs, all 61 sense codons are encoded in the genome sequence. The codon usage correlates well with the tRNA complement and is consistent with the high G+C content of the genome as the GC-rich codons predominate in the organism (Supplementary Table S1). Codons ATA (Ile), CGC (Arg), and CGA (Arg) as well as all codons that have a T at the third position, with the exception of CGT (Arg), do not appear to have a cognate tRNA in A. cellulolyticus. As a likely evolutionary adaptation to the available tRNAs for any given amino acid, codons that do not have a cognate tRNA occur with the least frequency in the A. cellulolyticus genome when compared to synonymous codons differing just in the 3rd position. However, the exceptions to these are for glycine (GGT (18.8%) > GGA (14.4%)), leucine (CTT (10.6%) > CTA (1.4%)), arginine (CGC (33.3%) > CGT (11.1%)), and valine (GTT (10.4%) > GTA (3.8%)). The relative preference for CGC codon over CGT codon follows the high G+C content of the genome, while the remaining four biases mentioned above may simply reflect evolutionary conservation of codon usage, as a similar trend is seen in Frankia (Supplementary Table S2). The functional significance of this bias remains elusive. The A. cellulolyticus genome encodes a parsimonious complement of 46 tRNAs. -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000725805Cyc: Streptomyces xanthophaeus Cellular Overview Connections between pathways are omitted for legibility. -

Riotinto: Un Universo De Mundos Microbianos
Ecosistemas 14 (2): 52-65. Mayo 2005. http://www.revistaecosistemas.net/articulo.asp?Id=110 Riotinto: un universo de mundos microbianos A. I. López-Archilla Departamento de Ecología. Facultad de Ciencias. Universidad Autónoma de Madrid. 28049 Cantoblanco, Madrid. España El río Tinto es un ambiente extremo caracterizado por un pH muy bajo y altas concentraciones de metales en disolución. Las extremas condiciones del río son en gran medida producidas y mantenidas por el componente biológico del ecosistema, principalmente por organismos procarióticos quimiolitótrofos. Las algas eucarióticas son los otros productores primarios del sistema mientras que hongos y bacterias heterotróficas juegan el papel de consumidores y descomponedores. Otros consumidores son protistas heterotróficos y, en ocasiones, rotíferos. El río en sí tiene una amplia variedad de ambientes en donde se desarrollan distintas comunidades (bentónicas en zonas aerobias, neustónicas en la superficie de aguas semiquietas, planctónicas en la columna de agua óxica y anóxica), además, las zonas adyacentes al río como los montones de mineral, las galerías mineras o el acuífero son ambientes distintos y aún inexplorados. A pesar de las condiciones extremas de este ecosistema, el río Tinto posee una gran diversidad de comunidades microbianas, cuyas interacciones son de gran interés para la ecología microbiana. The Tinto River is an extreme environment characterized by a very low pH and high concentration of heavy metals in solution. The extreme features of the river are produced and sustained by the biological components of the ecosystem, mainly by chemolithoautotrophic prokaryotic organisms. Eukaryotic algae are another primary producers in the system, while fungi and heterotrophic bacteria play the role of consumers and decomposers. -

Conservation of Core Complex Subunits Shaped the Structure And
University of Groningen Conservation of core complex subunits shaped the structure and function of photosystem I in the secondary endosymbiont alga Nannochloropsis gaditana Alboresi, Alessandro; Le Quiniou, Clotilde; Yadav, Sathish K N; Scholz, Martin; Meneghesso, Andrea; Gerotto, Caterina; Simionato, Diana; Hippler, Michael; Boekema, Egbert J.; Croce, Roberta Published in: New Phytologist DOI: 10.1111/nph.14156 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2017 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Alboresi, A., Le Quiniou, C., Yadav, S. K. N., Scholz, M., Meneghesso, A., Gerotto, C., ... Morosinotto, T. (2017). Conservation of core complex subunits shaped the structure and function of photosystem I in the secondary endosymbiont alga Nannochloropsis gaditana. New Phytologist, 213, 714-726. [nph.14156]. https://doi.org/10.1111/nph.14156 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum.