CYANIDE DEGRADATION by Bacillus Pumilus Cl: CELLULAR and MOLECULAR CHARACTERIZATION
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genomes of Polyextremophilic Cyanidiales Contain 1% 2 Horizontally Transferred Genes with Diverse Adaptive Functions 3 4 Alessandro W
bioRxiv preprint doi: https://doi.org/10.1101/526111; this version posted January 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 The genomes of polyextremophilic Cyanidiales contain 1% 2 horizontally transferred genes with diverse adaptive functions 3 4 Alessandro W. Rossoni1#, Dana C. Price2, Mark Seger3, Dagmar Lyska1, Peter Lammers3, 5 Debashish Bhattacharya4 & Andreas P.M. Weber1* 6 7 1Institute of Plant Biochemistry, Cluster of Excellence on Plant Sciences (CEPLAS), Heinrich 8 Heine University, Universitätsstraße 1, 40225 Düsseldorf, Germany 9 2Department of Plant Biology, Rutgers University, New Brunswick, NJ 08901, USA 10 3Arizona Center for Algae Technology and Innovation, Arizona State University, Mesa, AZ 11 85212, USA 12 4Department of Biochemistry and Microbiology, Rutgers University, New Brunswick, NJ 13 08901, USA 14 15 *Corresponding author: Prof. Dr. Andreas P.M. Weber, 16 e-mail: [email protected] 17 bioRxiv preprint doi: https://doi.org/10.1101/526111; this version posted January 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 18 Abstract 19 The role and extent of horizontal gene transfer (HGT) in eukaryotes are hotly disputed topics 20 that impact our understanding regarding the origin of metabolic processes and the role of 21 organelles in cellular evolution. -

The Cyanide Hydratase from Neurospora Crassa Forms a Helix Which Has a Dimeric Repeat
Appl Microbiol Biotechnol (2009) 82:271–278 DOI 10.1007/s00253-008-1735-4 BIOTECHNOLOGICALLY RELEVANT ENZYMES AND PROTEINS The cyanide hydratase from Neurospora crassa forms a helix which has a dimeric repeat Kyle C. Dent & Brandon W. Weber & Michael J. Benedik & B. Trevor Sewell Received: 2 July 2008 /Revised: 24 September 2008 /Accepted: 25 September 2008 / Published online: 23 October 2008 # Springer-Verlag 2008 Abstract The fungal cyanide hydratases form a functionally Introduction specialized subset of the nitrilases which catalyze the hydrolysis of cyanide to formamide with high specificity. The generation of cyanide containing waste results from These hold great promise for the bioremediation of cyanide cyanide utilization in a variety of applications, spanning wastes. The low resolution (3.0 nm) three-dimensional such diverse industries as gold and silver mining, metal reconstruction of negatively stained recombinant cyanide electroplating, polymer synthesis, and the production of hydratase fibers from the saprophytic fungus Neurospora fine chemicals, pharmaceuticals, dyes, and agricultural crassa by iterative helical real space reconstruction reveals products (Baxter and Cummings 2006). Traditionally, that enzyme fibers display left-handed D1 S5.4 symmetry chemical oxidation or stabilizations are used to treat these with a helical rise of 1.36 nm. This arrangement differs from wastes; however, many groups have been studying the previously characterized microbial nitrilases which demon- potential for bioremediation of such cyanide waste strate a structure built along similar principles but with a (O’Reilly and Turner 2003; Jandhyala et al. 2005). reduced helical twist. The cyanide hydratase assembly is The most promising approach utilizes cyanide degrada- stabilized by two dyadic interactions between dimers across tion by enzymes belonging to the nitrilase family (Pace and the one-start helical groove. -

Cyanide-Degrading Enzymes for Bioremediation A
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Texas A&M University CYANIDE-DEGRADING ENZYMES FOR BIOREMEDIATION A Thesis by LACY JAMEL BASILE Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE August 2008 Major Subject: Microbiology CYANIDE-DEGRADING ENZYMES FOR BIOREMEDIATION A Thesis by LACY JAMEL BASILE Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Approved by: Chair of Committee, Michael Benedik Committee Members, Susan Golden James Hu Wayne Versaw Head of Department, Vincent Cassone August 2008 Major Subject: Microbiology iii ABSTRACT Cyanide-Degrading Enzymes for Bioremediation. (August 2008) Lacy Jamel Basile, B.S., Texas A&M University Chair of Advisory Committee: Dr. Michael Benedik Cyanide-containing waste is an increasingly prevalent problem in today’s society. There are many applications that utilize cyanide, such as gold mining and electroplating, and these processes produce cyanide waste with varying conditions. Remediation of this waste is necessary to prevent contamination of soils and water. While there are a variety of processes being used, bioremediation is potentially a more cost effective alternative. A variety of fungal species are known to degrade cyanide through the action of cyanide hydratases, a specialized subset of nitrilases which hydrolyze cyanide to formamide. Here I report on previously unknown and uncharacterized nitrilases from Neurospora crassa, Gibberella zeae, and Aspergillus nidulans. Recombinant forms of four cyanide hydratases from N. -
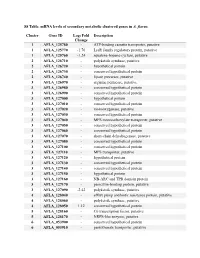
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

Comparative Genomics of Bradyrhizobium Japonicum CPAC
Siqueira et al. BMC Genomics 2014, 15:420 http://www.biomedcentral.com/1471-2164/15/420 RESEARCH ARTICLE Open Access Comparative genomics of Bradyrhizobium japonicum CPAC 15 and Bradyrhizobium diazoefficiens CPAC 7: elite model strains for understanding symbiotic performance with soybean Arthur Fernandes Siqueira1,2†, Ernesto Ormeño-Orrillo3†,RangelCelsoSouza4,ElisetePainsRodrigues5, Luiz Gonzaga Paula Almeida4, Fernando Gomes Barcellos5, Jesiane Stefânia Silva Batista6, Andre Shigueyoshi Nakatani2, Esperanza Martínez-Romero3, Ana Tereza Ribeiro Vasconcelos4 and Mariangela Hungria1,2* Abstract Background: The soybean-Bradyrhizobium symbiosis can be highly efficient in fixing nitrogen, but few genomic sequences of elite inoculant strains are available. Here we contribute with information on the genomes of two commercial strains that are broadly applied to soybean crops in the tropics. B. japonicum CPAC 15 (=SEMIA 5079) is outstanding in its saprophytic capacity and competitiveness, whereas B. diazoefficiens CPAC 7 (=SEMIA 5080) is known for its high efficiency in fixing nitrogen. Both are well adapted to tropical soils. The genomes of CPAC 15 and CPAC 7 were compared to each other and also to those of B. japonicum USDA 6T and B. diazoefficiens USDA 110T. Results: Differences in genome size were found between species, with B. japonicum having larger genomes than B. diazoefficiens. Although most of the four genomes were syntenic, genome rearrangements within and between species were observed, including events in the symbiosis island. In addition to the symbiotic region, several genomic islands were identified. Altogether, these features must confer high genomic plasticity that might explain adaptation and differences in symbiotic performance. It was not possible to attribute known functions to half of the predicted genes. -

Metatranscriptomic Analysis of Community Structure And
School of Environmental Sciences Metatranscriptomic analysis of community structure and metabolism of the rhizosphere microbiome by Thomas Richard Turner Submitted in partial fulfilment of the requirement for the degree of Doctor of Philosophy, September 2013 This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with the author and that use of any information derived there from must be in accordance with current UK Copyright Law. In addition, any quotation or extract must include full attribution. i Declaration I declare that this is an account of my own research and has not been submitted for a degree at any other university. The use of material from other sources has been properly and fully acknowledged, where appropriate. Thomas Richard Turner ii Acknowledgements I would like to thank my supervisors, Phil Poole and Alastair Grant, for their continued support and guidance over the past four years. I’m grateful to all members of my lab, both past and present, for advice and friendship. Graham Hood, I don’t know how we put up with each other, but I don’t think I could have done this without you. Cheers Salt! KK, thank you for all your help in the lab, and for Uma’s biryanis! Andrzej Tkatcz, thanks for the useful discussions about our projects. Alison East, thank you for all your support, particularly ensuring Graham and I did not kill each other. I’m grateful to Allan Downie and Colin Murrell for advice. For sequencing support, I’d like to thank TGAC, particularly Darren Heavens, Sophie Janacek, Kirsten McKlay and Melanie Febrer, as well as John Walshaw, Mark Alston and David Swarbreck for bioinformatic support. -

Purification and Characterization of a Novel Nitrilase of Rhodococcus
JOURNAL OF BACTERIOLOGY, Sept. 1990, p. 4807-4815 Vol. 172, No. 9 0021-9193/90/094807-09$02.00/0 Copyright X3 1990, American Society for Microbiology Purification and Characterization of a Novel Nitrilase of Rhodococcus rhodochrous K22 That Acts on Aliphatic Nitriles MICHIHIKO KOBAYASHI,* NORIYUKI YANAKA, TORU NAGASAWA, AND HIDEAKI YAMADA Department ofAgricultural Chemistry, Faculty ofAgriculture, Kyoto University, Sakyo-ku, Kyoto 606, Japan Received 18 April 1990/Accepted 8 June 1990 A novel nitrilase that preferentially catalyzes the hydrolysis of aliphatic nitriles to the corresponding carboxylic acids and ammonia was found in the cells of a facultative crotononitrile-utilizing actinomycete isolated from soil. The strain was taxonomically studied and identified as Rhodococcus rhodochrous. The nitrilase was purified, with 9.08% overall recovery, through five steps from a cell extract of the stain. After the last step, the purified enzyme appeared to be homogeneous, as judged by polyacrylamide gel electrophoresis, analytical centrifugation, and double immunodiffusion in agarose. The relative molecular weight values for the native enzyme, estimated from the ultracentrifugal equilibrium and by high-performance liquid chromatog- raphy, were approximately 604,000 + 30,000 and 650,000, respectively, and the enzyme consisted of 15 to 16 subunits identical in molecular weight (41,000). The enzyme acted on aliphatic olefinic nitriles such as crotononitrile and acrylonitrile as the most suitable substrates. The apparent Km values for crotononitrile and acrylonitrile were 18.9 and 1.14 mM, respectively. The nitrilase also catalyzed the direct hydrolysis of saturated aliphatic nitriles, such as valeronitrile, 4-chlorobutyronitrile, and glutaronitrile, to the correspond- ing acids without the formation of amide intermediates. -

Advances in Enzyme Biotechnology Advances in Enzyme Biotechnology
Pratyoosh Shukla Brett I. Pletschke Editors Advances in Enzyme Biotechnology Advances in Enzyme Biotechnology Pratyoosh Shukla • Brett I. Pletschke Editors Advances in Enzyme Biotechnology Editors Pratyoosh Shukla Brett I. Pletschke Department of Microbiology Department of Biochemistry, Maharshi Dayanand University Microbiology and Biotechnology Rohtak, Haryana , India Rhodes University Grahamstown, South Africa ISBN 978-81-322-1093-1 ISBN 978-81-322-1094-8 (eBook) DOI 10.1007/978-81-322-1094-8 Springer New Delhi Heidelberg New York Dordrecht London Library of Congress Control Number: 2013945175 © Springer India 2013 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifi cally the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfi lms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. Exempted from this legal reservation are brief excerpts in connection with reviews or scholarly analysis or material supplied specifi cally for the purpose of being entered and executed on a computer system, for exclusive use by the purchaser of the work. Duplication of this publication or parts thereof is permitted only under the provisions of the Copyright Law of the Publisher’s location, in its current version, and permission for use must always be obtained from Springer. Permissions for use may be obtained through RightsLink at the Copyright Clearance Center. Violations are liable to prosecution under the respective Copyright Law. The use of general descriptive names, registered names, trademarks, service marks, etc. -

Purification and Characterization of Nitrilase from Fusarium Solani
Process Biochemistry 45 (2010) 1115–1120 Contents lists available at ScienceDirect Process Biochemistry journal homepage: www.elsevier.com/locate/procbio Purification and characterization of nitrilase from Fusarium solani IMI196840 Vojtechˇ Vejvoda a, David Kubácˇ a,Alzbˇ etaˇ Davidová a, Ondrejˇ Kaplan a, Miroslav Sulcˇ a,b, Ondrejˇ Svedaˇ a, Radka Chaloupková c, Ludmila Martínková a,∗ a Institute of Microbiology, Centre of Biocatalysis and Biotransformation, Academy of Sciences of the Czech Republic, Vídenskᡠ1083, CZ-142 20 Prague, Czech Republic b Department of Biochemistry, Faculty of Science, Charles University Prague, Hlavova 8, CZ-128 40 Prague, Czech Republic c Loschmidt Laboratories, Department of Experimental Biology and Centre of Biocatalysis and Biotransformation, Faculty of Science, Masaryk University, Kamenice 5/A4, CZ-625 00 Brno, Czech Republic article info abstract Article history: Nitrilase activity in Fusarium solani IMI196840 (approx. 1500 U l−1 of culture broth) was induced by 2- Received 11 November 2009 cyanopyridine. The enzyme was purified by a factor of 20.3 at a yield of 26.9%. According to gel filtration, Received in revised form 26 March 2010 the holoenzyme was an approx. 550-kDa homooligomer consisting of subunits with a molecular weight Accepted 30 March 2010 of approximately 40 kDa, as determined by SDS-PAGE. Mass spectrometry analysis of the tryptic frag- ments suggested a high similarity of this enzyme to the hypothetical CN hydrolases from Aspergillus Keywords: oryzae, Gibberella zeae, Gibberella moniliformis and Nectria haematococca. Circular dichroism and fluo- Fusarium solani rescence spectra indicated that secondary structure content and overall tertiary structure, respectively, Nitrilase were almost identical in nitrilases from F. -

Structures, Functions, and Mechanisms of Filament Forming Enzymes: a Renaissance of Enzyme Filamentation
Structures, Functions, and Mechanisms of Filament Forming Enzymes: A Renaissance of Enzyme Filamentation A Review By Chad K. Park & Nancy C. Horton Department of Molecular and Cellular Biology University of Arizona Tucson, AZ 85721 N. C. Horton ([email protected], ORCID: 0000-0003-2710-8284) C. K. Park ([email protected], ORCID: 0000-0003-1089-9091) Keywords: Enzyme, Regulation, DNA binding, Nuclease, Run-On Oligomerization, self-association 1 Abstract Filament formation by non-cytoskeletal enzymes has been known for decades, yet only relatively recently has its wide-spread role in enzyme regulation and biology come to be appreciated. This comprehensive review summarizes what is known for each enzyme confirmed to form filamentous structures in vitro, and for the many that are known only to form large self-assemblies within cells. For some enzymes, studies describing both the in vitro filamentous structures and cellular self-assembly formation are also known and described. Special attention is paid to the detailed structures of each type of enzyme filament, as well as the roles the structures play in enzyme regulation and in biology. Where it is known or hypothesized, the advantages conferred by enzyme filamentation are reviewed. Finally, the similarities, differences, and comparison to the SgrAI system are also highlighted. 2 Contents INTRODUCTION…………………………………………………………..4 STRUCTURALLY CHARACTERIZED ENZYME FILAMENTS…….5 Acetyl CoA Carboxylase (ACC)……………………………………………………………………5 Phosphofructokinase (PFK)……………………………………………………………………….6 -

(12) Patent Application Publication (10) Pub. No.: US 2008/0148432 A1 Abad (43) Pub
US 2008O148432A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0148432 A1 Abad (43) Pub. Date: Jun. 19, 2008 (54) TRANSGENIC PLANTS WITH ENHANCED Publication Classification AGRONOMIC TRAITS (51) Int. Cl. AOIH 5/00 (2006.01) CI2N 5/82 (2006.01) (76) Inventor: Mark Scott Abad, Webster Grove, CI2N 5/04 (2006.01) MO (US) (52) U.S. Cl. ......... 800/279: 800/281; 435/419,435/468; 8OO/320.1 Correspondence Address: (57)57 ABSTRACT MONSANTO COMPANY This invention provides transgenic plant cells with recombi 800 N. LINDBERGHBLVD, ATTENTION: GAIL nant DNA for expression of proteins that are useful for P. WUELLNER, IP PARALEGAL (E2NA) imparting enhanced agronomic trait(s) to transgenic crop ST. LOUIS MO 631.67 9 plants. This invention also provides transgenic plants and 9 progeny seed comprising the transgenic plant cells where the plants are selected for having an enhanced trait selected from the group of traits consisting of enhanced water use effi (21) Appl. No.: 11/374,300 ciency, enhanced cold tolerance, increased yield, enhanced nitrogen use efficiency, enhanced seed protein and enhanced seed oil. Also disclosed are methods for manufacturing trans (22) Filed: Dec. 21, 2005 genic seed and plants with enhanced traits. Patent Application Publication Jun. 19, 2008 Sheet 1 of 3 US 2008/0148432 A1 Figure 1. 41905 - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - 127 O2 - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -

UNIVERSITY of CALIFORNIA Los Angeles Cloud-Based Analysis And
UNIVERSITY OF CALIFORNIA Los Angeles Cloud-based Analysis and Integration of Proteomics and Metabolomics Datasets A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics by Jeong Ho (Howard) Choi 2019 © Copyright by Jeong Ho (Howard) Choi 2019 ABSTRACT OF THE DISSERTATION Cloud-based Analysis and Integration of Proteomics and Metabolomics Datasets by Jeong Ho (Howard) Choi Doctor of Philosophy in Bioinformatics University of California, Los Angeles, 2019 Professor Peipei Ping, Chair Our capabilities to define cardiovascular health and disease using highly multivariate “omics” datasets have substantially increased in recent years. Advances in acquisition technologies as well as bioinformatics methods have paved the way for ultimately resolving every biomolecule comprising various human “omes”. Understanding how different “omes” change and interact with one another temporally will ultimately unveil multi-omic molecular signatures that inform pathologic mechanisms, indicate disease phenotypes, and identify new therapeutic targets. Herein we describe a thesis project that creates novel, contemporary data science methods and workflows to extract temporal molecular signatures of disease from multi-omics analyses, and develops integrated omics knowledgebases for the cardiovascular community at-large. Chapter 1 provides an overview of cardiac physiology and pathophysiology involved in cardiac remodeling and heart failure (HF). A description of the systematic characterization of cardiac proteomes and metabolomes is included, including methodologies for multi-omics phenotyping. Finally, an overview of bioinformatics methods for driver molecule discovery is provided, discussing strategies for characterizing temporal patterns and conducting functional enrichment. ii Chapter 2 describes computational approaches to discern the oxidative posttranslational modification (O-PTM) proteome, an important factor in cardiac remodeling.