Dynamic Modelling of the Fate of DDT in Lake Maggiore: Preliminary Results
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lago Maggiore E Valli
Lago Maggiore e Valli www.ticino-tourism.ch Lago Maggiore e Valli de-Pinakothek in der Casa Rusca, in der die verschiedenen Legate und Schenkungen bedeutender Künstler zu bewundern sind, unter ihnen auch die Schenkung von Jean und Ascona Marguerite Arp. Locarno, zusammen mit dem kleineren und Wer die Umgebung von Locarno besucht, nahen Ascona, darf man ruhig als Perle des erlebt eine wunderbare Natur: Von der subtro- Lago Maggiore bezeichnen. In schöner Lage pischen Landschaft längs der Seeufer bis hin und begünstigt von einem milden Klima sowie zur alpinen Welt der Berggipfel rund herum. einer üppigen, subtropischen Pflanzenwelt, Mit dem Schiff, das von Locarno, längs dem rühmt sich diese Kleinstadt einer touristischen Gambarogno und dem Linken Ufer des Lago Tradition, die auf die Zeit der Römer zurückgeht. Maggiore entlang fährt, erreicht man die Während des Filmfestivals, das Locarno herrlichen Brissagoinseln. Dort schmückt sich zu internationaler Bekanntheit verholfen hat, im Frühlung der berühmte botanische Park, Ziel verwandelt sich die Piazza Grande in einen zahlreicher Touristinnen und Touristen, mit offenen Saal, wie er wohl auf der Welt seines- Tausenden von schillernden Farben, die aus gleichen sucht. Und sie bietet viele weitere den grünen Wassern des Sees hervorstechen kulturelle und künstlerische Alternativen. wie ein wertvolles Schmuckstück. Fährt man Allein der Besuch der Altstadt, mit ihren dem rechten Ufer entlang, kann man Brissago typischen Restaurants und unzähligen kleinen und Ronco bewundern, um schliesslich in Läden mit den unterschiedlichsten Waren, Ascona an Land zu gehen. Das hübsche würde bereits eine Reise rechtfertigen. Aber Städtchen seit jeher Ziel von Künstlern und ebenso wunderbar ist ein Ausflug auf Cardada, Intellektuellen internationalen Rufs, beherbergt, den Berg mit seinen waldreichen Abhängen, in seinen Gässchen Museen und Kunstgalerien, die Locarno umgeben. -

Franklin University Switzerland Student Handbook 2020-2021
STUDENT HANDBOOK 2021-2022 ACADEMIC YEAR MISSION The Office of Student Life facilitates student learning and development through intercultural opportunities, immigration support, health and wellness services, and other co-curricular experiences. ABOUT US We know that student engagement outside the classroom is critical to success in the classroom and in life. Our programs and services are designed to help students achieve their academic goals; engage in experiential learning; develop intercultural maturity; cultivate relationships within Franklin and the surrounding communities; exhibit civic responsibility, and graduate with a stronger sense of cross-cultural perspectives that allows them to build careers that take them beyond national boundaries. EQUAL OPPORTUNITIES Franklin University Switzerland is committed to the principle of equal opportunity and to providing an academic and work environment free from discrimination. The University prohibits discrimination on the basis of race, color, national or ethnic origin, religion, sex, sexual orientation, gender identity or gender expression, age, disability, and other legally protected statuses. LEARNING OUTCOMES Students will be able to: • Demonstrate an understanding of, and develop relationships within, the Franklin and surrounding communities while utilizing Franklin and local community resources. • Demonstrate civic responsibility, ownership, and accountability on and off campus. • Display intercultural maturity in all aspects of your life. • Demonstrate the ability to contribute to -

Ticino on the Move
Tales from Switzerland's Sunny South Ticino on theMuch has changed move since 1882, when the first railway tunnel was cut through the Gotthard and the Ceneri line began operating. Mendrisio’sTHE LIGHT Processions OF TRADITION are a moving experience. CrystalsTREASURE in the AMIDST Bedretto THE Valley. ROCKS ChestnutsA PRICKLY are AMBASSADOR a fruit for all seasons. EasyRide: Travel with ultimate freedom. Just check in and go. New on SBB Mobile. Further information at sbb.ch/en/easyride. EDITORIAL 3 A lakeside view: Angelo Trotta at the Monte Bar, overlooking Lugano. WHAT'S NEW Dear reader, A unifying path. Sopraceneri and So oceneri: The stories you will read as you look through this magazine are scented with the air of Ticino. we o en hear playful things They include portraits of men and women who have strong ties with the local area in the about this north-south di- truest sense: a collective and cultural asset to be safeguarded and protected. Ticino boasts vide. From this year, Ticino a local rural alpine tradition that is kept alive thanks to the hard work of numerous young will be unified by the Via del people. Today, our mountain pastures, dairies, wineries and chestnut woods have also been Ceneri themed path. restored to life thanks to tourism. 200 years old but The stories of Lara, Carlo and Doris give off a scent of local produce: of hay, fresh not feeling it. milk, cheese and roast chestnuts, one of the great symbols of Ticino. This odour was also Vincenzo Vela was born dear to the writer Plinio Martini, the author of Il fondo del sacco, who used these words to 200 years ago. -
Buyers Seek a Coronavirus Escape in Italy's Lakes
HOME WORLD US COMPANIES TECH MARKETS GRAPHICS OPINION WORK & CAREERS LIFE & ARTS HOW TO SPEND IT Sign In Subscribe HOME WORLD US COMPANIES TECH MARKETS GRAPHICS OPINION WORK & CAREERS LIFE & ARTS HOW TO SPEND IT Sign In Subscribe CORONAVIRUS BUSINESS UPDATE Get 30 days’ complimentary access to our Coronavirus Business Get the newsletter now Update newsletter Latest on Prime property Architects embrace cabin fever Five of the world’s best homes for The German housing-market Wealthy buyers snap up ‘safe sale with swimming pools exception haven’ private islands to flee pandemic Prime property Add to myFT Buyers seek a coronavirus escape in Italy’s lakes The area’s open spaces and romantic vistas appeal to both internationals and locals Save Six-bedroom villa at Stresa, Lake Maggiore, €4.9m through Engel & Völkers George Steer 3 HOURS AGO 1 The lotus flowers and dahlias are in full bloom in the botanical gardens of Villa Taranto in Verbania, on the shores of Lake Maggiore in northern Italy. In a normal July, the estate would attract 1,000 paying visitors a day — this year it is closer to 300. “The town is calm, keeping our eyes open,” says Giuliana Zanzola, the estate’s tourist manager. “But fewer ticket sales mean we soon won’t have the money to pay the gardeners and ourselves. It’s really bad.” The lake forms part of the border between Piedmont and Lombardy, two regions that were badly hit by the coronavirus outbreak in the early spring. Lombardy accounts for close to half of all Covid-related deaths in the country. -

The Residence Time of the Water in Lago Maggiore (N. Italy): First Results from an Eulerian-Lagrangian Approach
J. Limnol., 69(1): 15-28, 2010 DOI: 10.3274/JL10-69-1-02 The residence time of the water in Lago Maggiore (N. Italy): first results from an Eulerian-Lagrangian approach Leonardo CASTELLANO*, Walter AMBROSETTI1), Luigi BARBANTI1) and Angelo ROLLA1) Matec Modelli Matematici, Via Rondoni 11, 20146 Milano, Italy 1)CNR Institute for Ecosystem Study, Largo V. Tonolli 50, 28922 Verbania-Pallanza, Italy *e-mail corresponding author: [email protected] ABSTRACT The paper describes a numerical study for estimating the spatial distribution of the hydraulic residence time in Lago Maggiore. A 3D eulerian time-dependent CFD code has been applied under real conditions, taking into account the effects of the monthly mean values of the mass flow rates and temperatures of all the tributaries, mass flow rate of the Ticino effluent and meteorological, hydrogeological and limnological parameters available from the rich data-base of CNR-ISE (Pallanza). The velocity distributions from these simulations were used to compute the paths of a number of massless markers with different initial positions and so evaluate their residence times within the lake. The results presented here follow a two-year simulation and show encouraging agreement with the mechanisms of mixing and of deep water oxygenation revealed by recent limnological studies carried out at CNR-ISE. Further studies are in progress to improve the results and extend the research over a time period of at least four years. Key words: residence time, hydro-meteorological parameters, eulerian mathematical model, -

923.250 Decreto Esecutivo Concernente Le Zone Di Protezione Pesca 2019-2024
923.250 Decreto esecutivo concernente le zone di protezione pesca 2019-2024 (del 24 ottobre 2018) IL CONSIGLIO DI STATO DELLA REPUBBLICA E CANTONE TICINO visti – la legge federale sulla pesca del 21 giugno 1991 (LFSP) e la relativa ordinanza del 24 novembre 1993 (OLFP), – la legge cantonale sulla pesca e sulla protezione dei pesci e dei gamberi indigeni del 26 giugno 1996 e il relativo regolamento di applicazione del 15 ottobre 1996, in particolare l’art. 19, – la Convenzione tra la Confederazione Svizzera e la Repubblica Italiana per la pesca nelle acque italo-svizzere del 1° aprile 1989, in particolare l’art. 6, decreta: Art. 1 Per il periodo dal 1° gennaio 2019 al 31 dicembre 2024, sono istituite le seguenti zone di protezione. a) Zone di protezione permanenti nei corsi d’acqua, dove la pesca è vietata: 1. Riale di Golino: dalla confluenza con il fiume Melezza alla prima cascata a monte della strada cantonale. 2. Torrente Brima ad Arcegno: dalla confluenza con il riale «Mulin di Cioss» all’entrata del paese di Arcegno, nonché tutta la tratta a valle dello sbarramento presso i mulini Simona a Losone. 3. Torrente Ribo a Vergeletto: dal ponte in località Custiell (pto. 891, poco a monte di Vergeletto) al ponte in ferro in località Zardin. 4. Fiume Bavona a Bignasco-Cavergno: dal ponte della cantonale a Bignasco fino alla passerella di Cavergno. 5. Roggia della piscicoltura di Sonogno: tutto il corso d’acqua fino alla confluenza con il fiume Verzasca. 6. Ronge di Alnasca a Brione Verzasca: dalla confluenza con il fiume Verzasca alle sorgenti. -
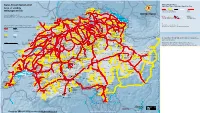
Swiss Travel System Map 2021
ai160326587010_STS-GB-Pass-S-21.pdf 1 21.10.20 09:37 Kruth Strasbourg | Paris Karlsruhe | Frankfurt | Dortmund | Hamburg | Berlin Stuttgart Ulm | München München Swiss Travel System 2021 Stockach Swiss Travel Pass Blumberg-Zollhaus Engen Swiss Travel Pass Youth | Swiss Travel Pass Flex Bargen Opfertshofen Überlingen Area of validity Seebrugg Beggingen Singen Ravensburg DEUTSCHLAND Radolfzell Schleitheim Hemmental Lines for unlimited travel (tunnel) Mulhouse Thayngen Mainau Geltungsbereich Meersburg Schaffhausen Ramsen Linien für unbegrenzte Fahrten (Tunnel) Zell (Wiesental) Wangen (Allgäu) Erzingen Oster- Neuhausen Stein a.R. Konstanz fingen Version/Stand/Etat/Stato:12.2020 (Baden) Rheinau Kreuzlingen Friedrichshafen Waldshut Due to lack of space not all lines are indicated. Subject to change. Marthalen Basel Weil a.R. Aus Platzgründen sind nicht alle Linien angegeben. Änderungen vorbehalten. Bad Zurzach Weinfelden Lines with reductions (50%, 1 25%) No reductions EuroAirport Riehen Koblenz Eglisau Frauenfeld Romanshorn Lindau Basel St.Johann Basel Möhlin Laufenburg Immenstadt Linien mit Vergünstigungen (50%, 1 25%) Keine Ermässigung Bad Bf Nieder- Stein-Säckingen Bülach Sulgen Arbon Basel Rheinfelden weningen Braunau Sonthofen Delle Pratteln Turgi Rorschach Bregenz Boncourt Ettingen Frick Brugg Zürich Bischofszell Rheineck Bonfol Liestal Baden Flughafen Winterthur Wil Rodersdorf Dornach Oberglatt Heiden St.Margrethen Aesch Gelterkinden Kloten Turbenthal St.Gallen Walzenhausen Roggenburg Wettingen Also valid for local public transport -

Inghams Guide and Insights to Lake Maggiore
Inghams guide and insights to Lake Maggiore Introduction to Lake Maggiore Lake Maggiore’s rich and colourful past seems ever present as you explore and enjoy this spectacular lake with shores bordered with lush Mediterranean vegetation, and the backdrop of the Alps in the distance. Ever since Napoleon built the Simplon highway from Geneva to Milan, which ran to the shores of Lake Maggiore, it became part of the Grand Tour, a haven for Europeans and British pioneers. Seduced by its Mediterranean climate, they built imposing villas and beautiful gardens attracting aristocrats, poets, writers and composers of the time. It was the setting for Ernest Hemingway’s “A Farewell to Arms” and it is easy to imagine his heroes fleeing from the Italian police in a rowing boat, trying to reach the Swiss border of the lake. Kings and Queens, writers, poets and composers such as Verdi, Wagner and Rossini have enjoyed the same panoramic views, nearby islands, exquisite botanical gardens, woodlands and tranquility that exist today. Lake Maggiore is the second largest lake on the south side of the Alps. The west bank is in the Italian region of Piedmont and the right in Lombardy. In the north Lake Maggiore crosses the Swiss border into the canton of Ticino. It is 65 km long and 10 km across at its widest point between Pallanza and Stresa. In spring and early summer the shores are lined with oleander and the scent of orange blossom pervades. Ever present are the magnificent palm trees and the heavenly scent of verbena. Across from Lake Maggiore lie the Borromean Islands and Isola Bella. -

Canton Ticino and the Italian Swiss Immigration to California
Swiss American Historical Society Review Volume 56 Number 1 Article 7 2020 Canton Ticino And The Italian Swiss Immigration To California Tony Quinn Follow this and additional works at: https://scholarsarchive.byu.edu/sahs_review Part of the European History Commons, and the European Languages and Societies Commons Recommended Citation Quinn, Tony (2020) "Canton Ticino And The Italian Swiss Immigration To California," Swiss American Historical Society Review: Vol. 56 : No. 1 , Article 7. Available at: https://scholarsarchive.byu.edu/sahs_review/vol56/iss1/7 This Article is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Swiss American Historical Society Review by an authorized editor of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Quinn: Canton Ticino And The Italian Swiss Immigration To California Canton Ticino and the Italian Swiss Immigration to California by Tony Quinn “The southernmost of Switzerland’s twenty-six cantons, the Ticino, may speak Italian, sing Italian, eat Italian, drink Italian and rival any Italian region in scenic beauty—but it isn’t Italy,” so writes author Paul Hofmann1 describing the one Swiss canton where Italian is the required language and the cultural tie is to Italy to the south, not to the rest of Switzerland to the north. Unlike the German and French speaking parts of Switzerland with an identity distinct from Germany and France, Italian Switzerland, which accounts for only five percent of the country, clings strongly to its Italian heritage. But at the same time, the Ticinese2 are fully Swiss, very proud of being part of Switzerland, and with an air of disapproval of Italy’s ever present government crises and its tie to the European Union and the Euro zone, neither of which Ticino has the slightest interest in joining. -

Lago Maggiore
Lago Maggiore Lago Maggiore (Verbano) Paese/i: Italia, Svizzera Regione/i: Ticino (CH), Piemonte (IT), Lombardia (IT) Provincia/e: Ticino: Distretto di Locarno Varese, Novara, Verbano-Cusio- Ossola Superficie: 212 km² Altitudine: 193 m s.l.m. Profondità 370 m massima: Immissari Ticino, Maggia, Toce, principali: Tresa Emissari Ticino principali: Bacino imbrifero: 6.599 km² « Se hai un cuore e una camicia, vendi la camicia e visita i dintorni del Lago Maggiore » (Stendhal) Il lago Maggiore o Verbano (indicato anche come lago di Locarno, Lach Magiur in dialetto lombardo occidentale) è un lago prealpino di origine glaciale, il secondo in Italia come superficie. Le sue rive sono condivise tra Svizzera (Canton Ticino) e Italia (province di Varese, Verbano- Cusio-Ossola e Novara). Morfologia Il lago Maggiore si trova ad un'altezza di circa 193 m s.l.m., la sua superficie è di 212 km² di cui circa l'80% è situata in territorio italiano e il rimanente 20% in territorio svizzero. Ha un perimetro di 170 km e una lunghezza di 54 km (la maggiore tra i laghi italiani); la larghezza massima è di 10 km e quella media di 3,9 km. Il volume d'acqua contenuto è pari a 37,5 miliardi di m³ di acqua con un tempo teorico di ricambio pari a circa 4 anni. Il bacino imbrifero è molto vasto, pari a circa 6.599 km² divisi quasi equamente tra Italia e Svizzera (il rapporto tra la superficie del bacino e quella del lago è pari 31,1), la massima altitudine di bacino è Punta Dufour nel massiccio del Monte Rosa (4.633 m s.l.m.) quella media è invece di 1.270 m s.l.m. -

The Maggia Cross-Fold : an Enigmatic Structure of the Lower Penninic Nappes of the Lepontine Alps
The Maggia cross-fold : an enigmatic structure of the Lower Penninic nappes of the Lepontine Alps Autor(en): Steck, Albrecht Objekttyp: Article Zeitschrift: Eclogae Geologicae Helvetiae Band (Jahr): 91 (1998) Heft 3 PDF erstellt am: 06.10.2021 Persistenter Link: http://doi.org/10.5169/seals-168427 Nutzungsbedingungen Die ETH-Bibliothek ist Anbieterin der digitalisierten Zeitschriften. Sie besitzt keine Urheberrechte an den Inhalten der Zeitschriften. Die Rechte liegen in der Regel bei den Herausgebern. Die auf der Plattform e-periodica veröffentlichten Dokumente stehen für nicht-kommerzielle Zwecke in Lehre und Forschung sowie für die private Nutzung frei zur Verfügung. Einzelne Dateien oder Ausdrucke aus diesem Angebot können zusammen mit diesen Nutzungsbedingungen und den korrekten Herkunftsbezeichnungen weitergegeben werden. Das Veröffentlichen von Bildern in Print- und Online-Publikationen ist nur mit vorheriger Genehmigung der Rechteinhaber erlaubt. Die systematische Speicherung von Teilen des elektronischen Angebots auf anderen Servern bedarf ebenfalls des schriftlichen Einverständnisses der Rechteinhaber. Haftungsausschluss Alle Angaben erfolgen ohne Gewähr für Vollständigkeit oder Richtigkeit. Es wird keine Haftung übernommen für Schäden durch die Verwendung von Informationen aus diesem Online-Angebot oder durch das Fehlen von Informationen. Dies gilt auch für Inhalte Dritter, die über dieses Angebot zugänglich sind. Ein Dienst der ETH-Bibliothek ETH Zürich, Rämistrasse 101, 8092 Zürich, Schweiz, www.library.ethz.ch http://www.e-periodica.ch -

Commissione Internazionale Per La Protezione Delle Acque Italo-Svizzere
ISSN: 1013-8099 Commissione Internazionale per la protezione delle acque italo-svizzere Ricerche sull'evoluzione del Lago Maggiore Aspetti limnologici Programma quinquennale 1998 – 2002 Campagna 2002 e Rapporto quinquennale 1998 - 2002 a cura di Roberto Bertoni Consiglio Nazionale delle Ricerche Istituto per lo Studio degli Ecosistemi Sezione di Idrobiologia ed Ecologia delle Acque Interne Verbania Pallanza I dati riportati nel presente volume possono essere utilizzati purché se ne citi la fonte come segue: C.N.R.-I.S.E. Sezione di Idrobiologia ed Ecologia delle Acque Interne- 2004. Ricerche sull'evoluzione del Lago Maggiore. Aspetti limnologici. Programma quinquennale 1998-2002. Campagna 2002. Commissione Internazionale per la protezione delle acque italo-svizzere (Ed.): 153 pp. ISSN: 1013-8099 Commissione Internazionale per la protezione delle acque italo-svizzere Ricerche sull'evoluzione del Lago Maggiore Aspetti limnologici Programma quinquennale 1998 - 2002 Campagna 2002 e Rapporto quinquennale 1998 - 2002 a cura di Roberto Bertoni Consiglio Nazionale delle Ricerche Istituto per lo Studio degli Ecosistemi Sezione di Idrobiologia ed Ecologia delle Acque Interne Verbania Pallanza RIASSUNTO Vengono qui riportati i risultati ottenuti dalle ricerche sul Lago Maggiore realizzate dalla Sezione di Idrobiologia ed Ecologia delle Acque Interne del CNR-ISE (già Istituto Italiano di Idrobiologia) per conto della Commissione Internazionale per la Protezione delle Acque Italo-Svizzere. Trattandosi dell’ultimo anno del quinto ciclo quinquennale, vengono anche illustrate le tendenze evolutive emerse nel corso del quinquennio, con- frontate con quanto osservato negli ultimi 50 anni. I risultati ottenuti evidenziano che la tendenza del lago ad evolvere verso una condi- zione di oligotrofia, già osservata nel precedente quinquennio, può essere confermata.