6 Chromosome Chapter
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genetical Society of Great Britain
Heredity 59 (1987) 151—160 The Genetical Society of Great Britain THEGENETICAL SOCIETY (Abstracts of papers presented at the TVVO HUNDRED AND FIFTH MEETING of the Society held on Friday, 14th and Saturday, 15th November 1986 at UNIVERSITY COLLEGE, LONDON) 1. Selection of somatic cell D. J. Porteous, P. A. Boyd, N. D. Hastie and hybrids with specific chromosome V. van Heyningen content for mapping the WAGR MAC Clinical and Population Cytogenetics Unit, Western General Hospital, Crewe Road, syndrome Edinburgh EH4 2XU. J. M. Fletcher, H. Morrison, J. A. Fantes, Clonedprobes for a number of available chromo- A. Seawright, S. Christie, D. J. Porteous, some ii assigned genes were used to define the N. D. Hastie and V. van Heyningen extent of deletions associated with the Wilms' MAC Clinical and Population Cytogenetics Unit, tumour, aniridia, genitourinary abnormalities and Western General Hospital, Crewe Road, mental retardation (WAGR) syndrome. Establish- Edinburgh EH4 2XU. ing reliable dosage studies for a number of different probes has proved difficult. We have therefore WAGR(Wilms tumour, aniridia, genitourinary abnormalities and mental retardation) syndrome concentrated on segregating the deleted chromo- is frequently associated with deletions on the short some 11 from a number of patients in somatic cell arm of chromosome 11. The deletions vary in size hybrids and analysing DNA from these to produce but always include part of band lipl3. To home a consistent map of chromosome lip. At the same in on the Wilms tumour and aniridia loci the end time we have determined the deletion breakpoints points of the different deletion breakpoints need at a molecular level and shown that the results are to be defined at the DNA level. -

Duodenal Webs: an Experience with 18 Patients
Journal of Neonatal Surgery 2012;1(2):20 O R I G I N A L A R T I C L E DUODENAL WEBS: AN EXPERIENCE WITH 18 PATIENTS Yogesh Kumar Sarin,* Akshay Sharma, Shalini Sinha, Vidyanand Pramod Deshpande Department of Pediatric Surgery, Maulana Azad Medical College, New Delhi-110002 * Corresponding Author Available at http://www.jneonatalsurg.com This work is licensed under a Creative Commons Attribution 3.0 Unported License How to cite: Sarin YK, Sharma A, Sinha S, Deshpande VP. Duodenal webs: an experience with 18 patients. J Neonat Surg 2012; 1: 20 ABSTRACT Aim: To describe the management and outcome of patients with duodenal webs, managed over a peri- od of 12 ½ years in our unit. Methods: It is a retrospective case series of 18 patients with congenital duodenal webs, managed in our unit, between 1999 and 2011. The medical record of these patients was retrieved and analyzed for demographic details, clinical presentation, associated anomalies, and outcome. Results: The median age of presentation was 8 days (range 1 day to 1.5 years). Antenatal diagnosis was made in only 2 (11.1%) patients. The commonest presentation was bilious vomiting. Associated anomalies were present in 8/18 patients, common being malrotation of gut. Down’s syndrome was seen in 2 patients and congenital heart disease in 1 patient. One patient had double duodenal webs. There was a delay in presentation of more than 5 days of life in 11/18 (61%) patients. Three patients who presented beyond neonatal age group had fenestrated duodenal membranes causing partial ob- struction. -

Genetic Syndromes and Genes Involved
ndrom Sy es tic & e G n e e n G e f Connell et al., J Genet Syndr Gene Ther 2013, 4:2 T o Journal of Genetic Syndromes h l e a r n a DOI: 10.4172/2157-7412.1000127 r p u y o J & Gene Therapy ISSN: 2157-7412 Review Article Open Access Genetic Syndromes and Genes Involved in the Development of the Female Reproductive Tract: A Possible Role for Gene Therapy Connell MT1, Owen CM2 and Segars JH3* 1Department of Obstetrics and Gynecology, Truman Medical Center, Kansas City, Missouri 2Department of Obstetrics and Gynecology, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania 3Program in Reproductive and Adult Endocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland, USA Abstract Müllerian and vaginal anomalies are congenital malformations of the female reproductive tract resulting from alterations in the normal developmental pathway of the uterus, cervix, fallopian tubes, and vagina. The most common of the Müllerian anomalies affect the uterus and may adversely impact reproductive outcomes highlighting the importance of gaining understanding of the genetic mechanisms that govern normal and abnormal development of the female reproductive tract. Modern molecular genetics with study of knock out animal models as well as several genetic syndromes featuring abnormalities of the female reproductive tract have identified candidate genes significant to this developmental pathway. Further emphasizing the importance of understanding female reproductive tract development, recent evidence has demonstrated expression of embryologically significant genes in the endometrium of adult mice and humans. This recent work suggests that these genes not only play a role in the proper structural development of the female reproductive tract but also may persist in adults to regulate proper function of the endometrium of the uterus. -

Diagnosis and Treatment of Jejunoileal Atresia
World J. Surg. 17, 310-3! 7, 1993 WORLD Journal of SURGERY 1993 by the Soci›233 O Internationale de Chirurgie Diagnosis and Treatment of Jejunoileal Atresia Robert J. Touloukian, M.D. Department of Surgery, Section of Pediatric Surgery, Yale University School of Medicine, and the Yale-New Haven Hospital, New Haven, Connecticut, U.S.A. A total of 116 cases of intestinal atresia or stenosis were encountered at the Classification Yale-New Haven Hospital between 1970 and 1990. Sites involved were the duodenum (n = 61; 53%), jejunum or ileum (n = 47; 46%), and colon (n Duodenum = 8; 7%). Ail but two patients underwent operative correction, for an overall survival rate of 92 %. Challenging problems were the management Sixty-one patients with duodenal atresia or stenosis were en- of apple-peel atresia (rive patients), multiple intestinal atresia with countered, including 12 with preampullary duodenal obstruc- short-gut syndrome (eight patients), and proximal jejunal atresia with megaduodenum requiring imbrication duodenoplasty (four patients). tion based on the absence of bile in the gastric contents. A Major assets in the improved outlook for intestinal atresia are prenatal diaphragm causing partial obstruction or duodenal stenosis was diagnosis, regionalization of neonatal care, improved recognition of found in 14 patients. An unusual cause of obstruction is associated conditions, innovative surgical methods, and uncomplicated complete absence of a duodenal segment accompanied by a long-terre total parenteral nutrition. mesenteric defect--seen in rive patients. Detecting a "wind- sock" web is critical because there is a tendency to confuse it with distal duodenal obstruction and the frequent occurrence of Atresia is the m0st common cause of congenital intestinal an anomalous biliary duct entering along its medial margin [9, obstruction and accounts for about one-third of all cases of 10]. -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Beckwith's Syndrome) M
Postgrad Med J: first published as 10.1136/pgmj.46.533.162 on 1 March 1970. Downloaded from 162 Case reports VAN DER SLUYS VEER, J., CHOUFOER, J.C., QUERIDO, A., Lancet, i, 1416. VAN DER HEUL, R.O., HOLLANDER, C.F. & VAN RIJSSEL, ZOLLINGER, R.M. & ELLIOTT, D.W. (1959) Pancreatic T.G. (1964) Metastasizing islet cell tumour of the pancreas endocrine function and peptic ulceration. Gastroenter- associated with hypoglycaemia and carcinoid syndrome. ology, 37, 401. Macroglossia, abnormal umbilicus and hypoglycaemia (Beckwith's syndrome) M. W. MONCRIEFF* J. R. MANN M.A., B.M., M.R.C.P. M.B., M.R.C.P., D.C.H. Lecturer in Paediatrics, Registrar in Paediatrics, University of Birmingham Birmingham Children's Hospital A. R. GOLDSMITH G. W. CHANCE M.B.B.S. M.B., M.R.C.P., D.C.H. Registrar in Pathology, Senior Lecturer in Paediatrics, Birmingham Children's Hospital University ofBirmingham Introduction lobe and three had a dome-like elevation of the The syndrome of exomphalos, macroglossia, post- posterior part of the diaphragm. The six surviving natal somatic gigantism and severe hypoglycaemia children developed a characteristic facies with prog- copyright. in various combinations was first described in seven nathos, mid-facial under development and slight infants by Beckwith (1963) and Beckwith et al. exophthalmos, and a mid-line frontal ridge. Post- (1964). At necropsy the main features were cyto- natal somatic gigantism occurred in five of the six megaly of the foetal adrenal cortex, renal medullary survivors. A further seven cases with macroglossia dysplasia, and hyperplasia of the pancreas and and umbilical abnormality were reported by Shafer kidneys. -

Chromosome 6 Abnormalities in Ovarian Surface Epithelial Tumors of Borderline Malignancy Suggest a Genetic Continuum in the Progression Model of Ovarian Neoplasms1
3404 Vol. 7, 3404–3409, November 2001 Clinical Cancer Research Chromosome 6 Abnormalities in Ovarian Surface Epithelial Tumors of Borderline Malignancy Suggest a Genetic Continuum in the Progression Model of Ovarian Neoplasms1 Maria Grazia Tibiletti, Barbara Bernasconi, Conclusion: Alterations in the above-mentioned inter- Daniela Furlan, Paola Bressan, Roberta Cerutti, val are a common finding in advanced ovarian carcinomas but also in benign ovarian cysts, implying that some tumors Carla Facco, Massimo Franchi, Cristina Riva, of borderline malignancy may arise from benign tumors and Raffaella Cinquetti, Carlo Capella, and that malignant ones may evolve from tumors of borderline Roberto Taramelli2 malignancy. Genes located in 6q27 seem to be crucial for this Laboratorio di Anatomia Patologica Ospedale di Circolo, Fondazione mechanism of early events in ovarian tumorigenesis. Macchi, 21100 Varese [M. G. T., C. F., C. R.]; and Dipartimento di Scienze Cliniche e Biologiche [B. B., D. F., P. B., R. Ce., C. C.], INTRODUCTION Clinica Ostetrica e Ginecologica [M. F.], and Dipartimento di Biologia Strutturale e Funzionale [R. Ci., R. T.], Universita` Ovarian cancer represents the most lethal of the gyneco- dell’Insubria, 21100 Varese, Italy logical tumors, and its pathogenesis is poorly understood. More than 80% of malignant tumors of the ovary are of epithelial origin and arise from the surface epithelium (1). Ovarian surface ABSTRACT epithelial tumors are divided into three histological subtypes: Purpose: We used conventional cytogenetics, molecular benign cystoadenoma, TBM,3 and AC, which are related to a cytogenetics, and molecular genetic analyses to study the different biological behavior (1). The molecular events under- pattern of allelic loss on chromosome 6q in a cohort of lying the development of ovarian tumorigenesis are still largely borderline epithelial ovarian tumors. -

Clinical Teaching Unit 3
Queen’s University Department of Surgery – Division of General Surgery Rotation Specific Goals and Objectives Clinical Teaching Unit #3 (CTU#3) – Pediatric Surgery Rotation Description The service of pediatric surgery is part of CTU #3. The service is comprised of Drs. Kolar and Winthrop. Dr. Kolar and Dr. Winthrop are available (as per the call schedule) for all surgical patients less than the age of 18 years from 8 am until 5pm. After 5pm the pediatric surgeon is on call for all patients 12 and under. The pediatric surgeons are also available for consult by the surgical residents or adult surgeon on call at any time. If a patient less than the age of 18 years is taken to the OR for trauma, congenital issues or any surgical issue that is complex the pediatric surgeons would like to be contacted. A rotation in Pediatric Surgery will give residents the opportunity to become familiar with the unique needs of infants, children and adolescents as surgical patients. Some of the surgical diseases encountered in children and adolescents are similar in their presentation, management and outcome with their adult counterparts; others are quite different. The fundamental principles of surgical care, however, are similar to those that govern surgical practice in other age groups. Goals: 1. Define the principles of investigation and management of infants, children and adolescents requiring surgical treatment. 2. Gain practical experience in the assessment, management, and indications for surgical treatment of common paediatric conditions. 3. Learn to perform certain paediatric surgical procedures. 4. Learn the principles of decision-making regarding the timing of surgery for infants, children and adolescents with complex paediatric surgical problems, including the preparation and transport to a paediatric surgical centre for neonates requiring correction of congenital anomalies. -

Preoperative Evaluation and Comprehensive Risk Assessment For
Pediatric Anesthesia ISSN 1155-5645 SPECIAL INTEREST ARTICLE (REVIEW) Preoperative evaluation and comprehensive risk assessment for children with Down syndrome Amy Feldman Lewanda1, Andrew Matisoff2, Mary Revenis3, Ashraf Harahsheh4, Craig Futterman5, Gustavo Nino6, Jay Greenberg7, John S. Myseros8, Kenneth N. Rosenbaum1 & Marshall Summar1 1 Division of Genetics & Metabolism, Children’s National Health System, Washington, DC, USA 2 Divisions of Anesthesiology, Sedation, and Perioperative Medicine, Children’s National Health System, Washington, DC, USA 3 Division of Neonatology, Children’s National Health System, Washington, DC, USA 4 Division of Cardiology, Children’s National Health System, Washington, DC, USA 5 Division of Critical Care Medicine, Children’s National Health System, Washington, DC, USA 6 Divisions of Pulmonary Medicine and Sleep Medicine, Children’s National Health System, Washington, DC, USA 7 Divisions of Hematology and Oncology, Children’s National Health System, Washington, DC, USA 8 Division of Neurosurgery, Children’s National Health System, Washington, DC, USA Keywords Summary Down syndrome; trisomy 21; surgery; anesthesia; perioperative; preoperative Down syndrome is a common chromosome disorder affecting all body sys- tems. This creates unique physiologic concerns that can affect safety during Correspondence anesthesia and surgery. Little consensus exists, however, on the best way to Amy Feldman Lewanda, Division of evaluate children with Down syndrome in preparation for surgery. We review Genetics & Metabolism, Children’s National a number of salient topics affecting these children in the perioperative period, Health System, 111 Michigan Ave. NW, including cervical spine instability, cardiovascular abnormalities, pulmonary Washington, DC 20010, USA Email: [email protected] hypertension, upper airway obstruction, hematologic disturbances, prematu- rity, low birth weight, and the use of supplements and alternative therapies. -
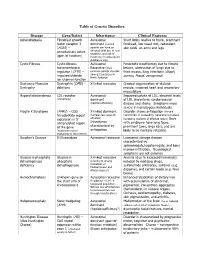
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical Features Achondroplasia Fibroblast growth Autosomal Short limbs relative to trunk, prominent factor receptor 3 dominant (normal forehead, low nasal root, redundant (FGR3) – parents can have an skin folds on arms and legs constitutively active affected child due to new mutation, and risk of (gain of function) recurrence in subsequent children is low) Cystic Fibrosis Cystic fibrosis Autosomal Pancreatic insufficiency due to fibrotic transmembrane Recessive (most lesions, obstruction of lungs due to regulator (CFTR) – common genetic disorder thick mucus, lung infections (Staph, impaired chloride among Caucasians in aureus, Pseud. aeruginosa) North America) ion channel function Duchenne Muscular Dystrophin (DMD) - X-linked recessive Gradual degeneration of skeletal Dystrophy deletions muscle, impaired heart and respiratory musculature Hypercholesterolemia LDL receptor Autosomal Impaired uptake of LDL, elevated levels (commonly) dominant of LDL cholesterol, cardiovascular (haploinsufficiency) disease and stroke. Symptoms more severe in homozygous individuals Fragile X Syndrome (FMR1) – CGG X-linked dominant Disorder shows anticipation (female trinucleotide repeat (females less severely transmitters in succeeding generations produce expansion in 5’ affected) increasing numbers of affected males) Boys untranslated region Inheritance with syndrome have long faces, of the gene characterized by prominent jaws, large ears, and are (expansion occurs anticipation likely to be mentally retarded. exclusively in the mother) Gaucher’s Disease Β-Glucosidase Autosomal recessive Lysosomal storage disease characterized by splenomegaly,hepatomegaly, and bone marrow infiltration. Neurological symptoms are not common Glucose 6-phosphate Glucose 6- X-linked recessive Anemia (due to increased hemolysis) dehydrogenase phosphate (prominent among induced by oxidizing drugs, deficiency dehydrogenase individuals of sulfonamide antibiotics, sulfones (e.g. -

PDF WEETH LECTURE Early Oral Cancers and Precancers
12/28/2018 CAUTION WEETH LECTUURE • Participants should be cautioned about the potential risks of using limited knowledge when integrating new techniques. EARLY ORAL CANCERS AND PRE-CANCERS MARY RIEPMA ROSS THEATER LINCOLN NEBRASKA JANUARY 4, 2019 DONALD M. COHEN DMD, MS, MBA PROFESSOR OF ORAL & MAXILOFACIAL PATHOLOGY ACTING CHAIR DEPARTMENT OF ORAL DIAGNOSTIC SCIENCS UNIVERSITY OF FLORIDA COLLEGE OF DENTISTRY GAINESVILLE, FLORIDA [email protected] 800-500-7585 Conflicts of Interests Course Objectives • Neither my immediate family nor I have any • Upon completion of this course, participants should be able to: financial interests that would create a conflict of • Recognize and formulate a differential diagnosis, understand the etiology and interest or restrict our independent judgment management of various oral and maxillofacial conditions. with regard to the content of this course. • Better recognize early mailignacies, improve diagnostic skills for oral soft and hard tissue lesions through practice sessions utilizing the audience response devices. GENERAL DENTIST TO ORAL SURGEON ORAL SURGOEN NOT TO WORRY PLEASE TAKE OUT TWO LOOSE TEETH TWO WEEK FOLLOW UP!! 1 12/28/2018 IDIOPATHIC LEUKOPLAKIA • FEATURES TO WORRY ABOUT • Occurrence in non-smoker • Thickened often corrugated appearance • Associated erythema • High risk location-horseshoe shaped area • ??pain • Multifocal or recurrent NON-SMOKERS LEUKOPLAKIA TONSILLAR CRYPT EPITHELIUM • Stratified squamous but basaloid so virus can • 5-8 times INCREASED risk of oral cancer invade -

Case Report Surgical Treatment of Congenital True Macroglossia
Hindawi Publishing Corporation Case Reports in Dentistry Volume 2013, Article ID 489194, 5 pages http://dx.doi.org/10.1155/2013/489194 Case Report Surgical Treatment of Congenital True Macroglossia Sabrina Araújo Pinho Costa, Mário César Pereira Brinhole, Rogério Almeida da Silva, Daniel Hacomar dos Santos, and Mayko Naruhito Tanabe DepartmentofOralandMaxillofacialSurgery,VilaPenteadoGeneralHospital,AvenueMinistroPetronioˆ Portela, 1642, Freguesia do O,´ 02802-120 Sao˜ Paulo, SP, Brazil Correspondence should be addressed to Sabrina Araujo´ Pinho Costa; [email protected] Received 26 August 2013; Accepted 20 October 2013 Academic Editors: P. Lopez Jornet, A. Mansourian, Y. Nakagawa, and P. I. Varela-Centelles Copyright © 2013 Sabrina Araujo´ Pinho Costa et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Macroglossia is a morphological and volumetric alteration of the tongue, caused by muscular hypertrophy, vascular malformation, metabolic diseases, and idiopathic causes and also associated with Down and Beckwith-Wiedemann syndromes. This alteration can cause dental-muscle-skeletal deformities, orthodontic instability, masticatory problems, and alterations in the taste and speech. In this paper we present a case of true macroglossia diagnosed in a female patient, 26 years, melanoderma, no family history of disease, with a history of relapse of orthodontic treatment for correction of open bite, loss of the lower central incisors, and complaint of difficulty in phonation. The patient was submitted to glossectomy under general anesthesia using the “keyhole” technique, with objective to provide reduction of the lingual length and width. The patient developed with good repair, without taste and motor alterations and discrete paresthesia at the apex of the tongue.