Supplementary Table 1. List of Biological Candidate Genes and Their Functional Roles for Genetic Variant Analysis 2 (N=153)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genetical Society of Great Britain
Heredity 59 (1987) 151—160 The Genetical Society of Great Britain THEGENETICAL SOCIETY (Abstracts of papers presented at the TVVO HUNDRED AND FIFTH MEETING of the Society held on Friday, 14th and Saturday, 15th November 1986 at UNIVERSITY COLLEGE, LONDON) 1. Selection of somatic cell D. J. Porteous, P. A. Boyd, N. D. Hastie and hybrids with specific chromosome V. van Heyningen content for mapping the WAGR MAC Clinical and Population Cytogenetics Unit, Western General Hospital, Crewe Road, syndrome Edinburgh EH4 2XU. J. M. Fletcher, H. Morrison, J. A. Fantes, Clonedprobes for a number of available chromo- A. Seawright, S. Christie, D. J. Porteous, some ii assigned genes were used to define the N. D. Hastie and V. van Heyningen extent of deletions associated with the Wilms' MAC Clinical and Population Cytogenetics Unit, tumour, aniridia, genitourinary abnormalities and Western General Hospital, Crewe Road, mental retardation (WAGR) syndrome. Establish- Edinburgh EH4 2XU. ing reliable dosage studies for a number of different probes has proved difficult. We have therefore WAGR(Wilms tumour, aniridia, genitourinary abnormalities and mental retardation) syndrome concentrated on segregating the deleted chromo- is frequently associated with deletions on the short some 11 from a number of patients in somatic cell arm of chromosome 11. The deletions vary in size hybrids and analysing DNA from these to produce but always include part of band lipl3. To home a consistent map of chromosome lip. At the same in on the Wilms tumour and aniridia loci the end time we have determined the deletion breakpoints points of the different deletion breakpoints need at a molecular level and shown that the results are to be defined at the DNA level. -

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

The Functions of DNA Damage Factor RNF8 in the Pathogenesis And
Int. J. Biol. Sci. 2019, Vol. 15 909 Ivyspring International Publisher International Journal of Biological Sciences 2019; 15(5): 909-918. doi: 10.7150/ijbs.31972 Review The Functions of DNA Damage Factor RNF8 in the Pathogenesis and Progression of Cancer Tingting Zhou 1, Fei Yi 1, Zhuo Wang 1, Qiqiang Guo 1, Jingwei Liu 1, Ning Bai 1, Xiaoman Li 1, Xiang Dong 1, Ling Ren 2, Liu Cao 1, Xiaoyu Song 1 1. Institute of Translational Medicine, China Medical University; Key Laboratory of Medical Cell Biology, Ministry of Education; Liaoning Province Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning Province, China 2. Department of Anus and Intestine Surgery, First Affiliated Hospital of China Medical University, Shenyang, Liaoning Province, China Corresponding authors: Xiaoyu Song, e-mail: [email protected] and Liu Cao, e-mail: [email protected]. Key Laboratory of Medical Cell Biology, Ministry of Education; Institute of Translational Medicine, China Medical University; Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning Province, 110122, China. Tel: +86 24 31939636, Fax: +86 24 31939636. © Ivyspring International Publisher. This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2018.12.03; Accepted: 2019.02.08; Published: 2019.03.09 Abstract The really interesting new gene (RING) finger protein 8 (RNF8) is a central factor in DNA double strand break (DSB) signal transduction. -

CRISPR Screening of Porcine Sgrna Library Identifies Host Factors
ARTICLE https://doi.org/10.1038/s41467-020-18936-1 OPEN CRISPR screening of porcine sgRNA library identifies host factors associated with Japanese encephalitis virus replication Changzhi Zhao1,5, Hailong Liu1,5, Tianhe Xiao1,5, Zichang Wang1, Xiongwei Nie1, Xinyun Li1,2, Ping Qian2,3, Liuxing Qin3, Xiaosong Han1, Jinfu Zhang1, Jinxue Ruan1, Mengjin Zhu1,2, Yi-Liang Miao 1,2, Bo Zuo1,2, ✉ ✉ Kui Yang4, Shengsong Xie 1,2 & Shuhong Zhao 1,2 1234567890():,; Japanese encephalitis virus (JEV) is a mosquito-borne zoonotic flavivirus that causes ence- phalitis and reproductive disorders in mammalian species. However, the host factors critical for its entry, replication, and assembly are poorly understood. Here, we design a porcine genome-scale CRISPR/Cas9 knockout (PigGeCKO) library containing 85,674 single guide RNAs targeting 17,743 protein-coding genes, 11,053 long ncRNAs, and 551 microRNAs. Subsequently, we use the PigGeCKO library to identify key host factors facilitating JEV infection in porcine cells. Several previously unreported genes required for JEV infection are highly enriched post-JEV selection. We conduct follow-up studies to verify the dependency of JEV on these genes, and identify functional contributions for six of the many candidate JEV- related host genes, including EMC3 and CALR. Additionally, we identify that four genes associated with heparan sulfate proteoglycans (HSPGs) metabolism, specifically those responsible for HSPGs sulfurylation, facilitate JEV entry into porcine cells. Thus, beyond our development of the largest CRISPR-based functional genomic screening platform for pig research to date, this study identifies multiple potentially vulnerable targets for the devel- opment of medical and breeding technologies to treat and prevent diseases caused by JEV. -

Mediator of DNA Damage Checkpoint Protein 1 Facilitates V(D)J Recombination in Cells Lacking DNA Repair Factor XLF
biomolecules Article Mediator of DNA Damage Checkpoint Protein 1 Facilitates V(D)J Recombination in Cells Lacking DNA Repair Factor XLF Carole Beck 1,2, Sergio Castañeda-Zegarra 1,2 , Camilla Huse 1,2, Mengtan Xing 1,2 and Valentyn Oksenych 1,2,3,* 1 Department of Clinical and Molecular Medicine (IKOM), Norwegian University of Science and Technology (NTNU), 7491 Trondheim, Norway; [email protected] (C.B.); [email protected] (S.C.-Z.); [email protected] (C.H.); [email protected] (M.X.) 2 St. Olavs Hospital, Trondheim University Hospital, Clinic of Medicine, Postboks 3250 Sluppen, 7006 Trondheim, Norway 3 Department of Biosciences and Nutrition (BioNuT), Karolinska Institutet, 14183 Huddinge, Sweden * Correspondence: [email protected] Received: 27 November 2019; Accepted: 24 December 2019; Published: 30 December 2019 Abstract: DNA double-strand breaks (DSBs) trigger the Ataxia telangiectasia mutated (ATM)-dependent DNA damage response (DDR), which consists of histone H2AX, MDC1, RNF168, 53BP1, PTIP, RIF1, Rev7, and Shieldin. Early stages of B and T lymphocyte development are dependent on recombination activating gene (RAG)-induced DSBs that form the basis for further V(D)J recombination. Non-homologous end joining (NHEJ) pathway factors recognize, process, and ligate DSBs. Based on numerous loss-of-function studies, DDR factors were thought to be dispensable for the V(D)J recombination. In particular, mice lacking Mediator of DNA Damage Checkpoint Protein 1 (MDC1) possessed nearly wild-type levels of mature B and T lymphocytes in the spleen, thymus, and bone marrow. NHEJ factor XRCC4-like factor (XLF)/Cernunnos is functionally redundant with ATM, histone H2AX, and p53-binding protein 1 (53BP1) during the lymphocyte development in mice. -

Combinatorial Genomic Data Refute the Human Chromosome 2 Evolutionary Fusion and Build a Model of Functional Design for Interstitial Telomeric Repeats
The Proceedings of the International Conference on Creationism Volume 8 Print Reference: Pages 222-228 Article 32 2018 Combinatorial Genomic Data Refute the Human Chromosome 2 Evolutionary Fusion and Build a Model of Functional Design for Interstitial Telomeric Repeats Jeffrey P. Tomkins Institute for Creation Research Follow this and additional works at: https://digitalcommons.cedarville.edu/icc_proceedings Part of the Biology Commons, and the Genomics Commons DigitalCommons@Cedarville provides a publication platform for fully open access journals, which means that all articles are available on the Internet to all users immediately upon publication. However, the opinions and sentiments expressed by the authors of articles published in our journals do not necessarily indicate the endorsement or reflect the views of DigitalCommons@Cedarville, the Centennial Library, or Cedarville University and its employees. The authors are solely responsible for the content of their work. Please address questions to [email protected]. Browse the contents of this volume of The Proceedings of the International Conference on Creationism. Recommended Citation Tomkins, J.P. 2018. Combinatorial genomic data refute the human chromosome 2 evolutionary fusion and build a model of functional design for interstitial telomeric repeats. In Proceedings of the Eighth International Conference on Creationism, ed. J.H. Whitmore, pp. 222–228. Pittsburgh, Pennsylvania: Creation Science Fellowship. Tomkins, J.P. 2018. Combinatorial genomic data refute the human chromosome 2 evolutionary fusion and build a model of functional design for interstitial telomeric repeats. In Proceedings of the Eighth International Conference on Creationism, ed. J.H. Whitmore, pp. 222–228. Pittsburgh, Pennsylvania: Creation Science Fellowship. COMBINATORIAL GENOMIC DATA REFUTE THE HUMAN CHROMOSOME 2 EVOLUTIONARY FUSION AND BUILD A MODEL OF FUNCTIONAL DESIGN FOR INTERSTITIAL TELOMERIC REPEATS Jeffrey P. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination
The Journal of Neuroscience, August 31, 2011 • 31(35):12579–12592 • 12579 Cellular/Molecular The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination Junji Yamauchi,1,3,5 Yuki Miyamoto,1 Hajime Hamasaki,1,3 Atsushi Sanbe,1 Shinji Kusakawa,1 Akane Nakamura,2 Hideki Tsumura,2 Masahiro Maeda,4 Noriko Nemoto,6 Katsumasa Kawahara,5 Tomohiro Torii,1 and Akito Tanoue1 1Department of Pharmacology and 2Laboratory Animal Resource Facility, National Research Institute for Child Health and Development, Setagaya, Tokyo 157-8535, Japan, 3Department of Biological Sciences, Tokyo Institute of Technology, Midori, Yokohama 226-8501, Japan, 4IBL, Ltd., Fujioka, Gumma 375-0005, Japan, and 5Department of Physiology and 6Bioimaging Research Center, Kitasato University School of Medicine, Sagamihara, Kanagawa 252-0374, Japan In development of the peripheral nervous system, Schwann cells proliferate, migrate, and ultimately differentiate to form myelin sheath. In all of the myelination stages, Schwann cells continuously undergo morphological changes; however, little is known about their underlying molecular mechanisms. We previously cloned the dock7 gene encoding the atypical Rho family guanine-nucleotide exchange factor (GEF) and reported the positive role of Dock7, the target Rho GTPases Rac/Cdc42, and the downstream c-Jun N-terminal kinase in Schwann cell migration (Yamauchi et al., 2008). We investigated the role of Dock7 in Schwann cell differentiation and myelination. Knockdown of Dock7 by the specific small interfering (si)RNA in primary Schwann cells promotes dibutyryl cAMP-induced morpholog- ical differentiation, indicating the negative role of Dock7 in Schwann cell differentiation. It also results in a shorter duration of activation of Rac/Cdc42 and JNK, which is the negative regulator of myelination, and the earlier activation of Rho and Rho-kinase, which is the positive regulator of myelination. -
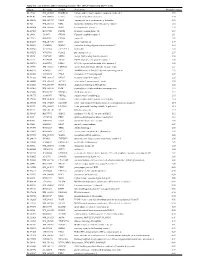
Supplementary Table 1
Table S1. List of Genes Differentially Expressed in YB-1 siRNA-Transfected MCF-7 Cells Unigene Accession Symbol Description Mean fold change Hs.17466 NM_004585 RARRES3 retinoic acid receptor responder (tazarotene induced) 3 3.57 Hs.64746 NM_004669 CLIC3 chloride intracellular channel 3 3.33 Hs.516155 NM_001747 CAPG capping protein (actin filament), gelsolin-like 2.88 Hs.926 NM_002463 MX2 myxovirus (influenza virus) resistance 2 (mouse) 2.72 Hs.643494 NM_005410 SEPP1 selenoprotein P, plasma, 1 2.70 Hs.267038 BC017500 POF1B premature ovarian failure 1B 2.67 Hs.20961 U63917 GPR30 G protein- coupled receptor 30 2532.53 Hs.199877 BE645967 CPNE4 copine IV 2.49 Hs.632177 NM_017458 MVP major vault protein 2.48 Hs.528836 AA005023 NOD27 nucleotide-binding oligomerization domains 27 2.43 Hs.360940 AL589866 dJ222E13.1 kraken-like 2.40 Hs.515575 AI743780 PLAC2 placenta-specific 2 2.37 Hs.25674 AI827820 MBD2 methyl-CpG binding domain protein 2 2.36 Hs.12229 AA149594 TIEG2 TGFB inducible early growth response 2 2.33 Hs.482730 AA053711 EDIL3 EGF-like repeats and discoidin I-like domains 3 2.32 Hs. 370771 NM_000389 CDKN1A cyclin- depen dent kinase in hibitor 1A (p 21, Cip 1) 2282.28 Hs.469175 AI341537 JFC1 NADPH oxidase-related, C2 domain-containing protein 2.28 Hs.514821 AF043341 CCL5 chemokine (C-C motif) ligand 5 2.27 Hs.591292 NM_023915 GPR87 G protein-coupled receptor 87 2.26 Hs.500483 NM_001613 ACTA2 actin, alpha 2, smooth muscle, aorta 2.24 Hs.632824 NM_006729 DIAPH2 diaphanous homolog 2 (Drosophila) 2.22 Hs.369430 NM_000919 PAM peptidylglycine -

Assembly Factors for the Membrane Arm of Human Complex I
Assembly factors for the membrane arm of human complex I Byron Andrews, Joe Carroll, Shujing Ding, Ian M. Fearnley, and John E. Walker1 Medical Research Council Mitochondrial Biology Unit, Cambridge CB2 0XY, United Kingdom Contributed by John E. Walker, October 14, 2013 (sent for review September 12, 2013) Mitochondrial respiratory complex I is a product of both the nuclear subunits in a fungal enzyme from Yarrowia lipolytica seem to be and mitochondrial genomes. The integration of seven subunits distributed similarly (12, 13). encoded in mitochondrial DNA into the inner membrane, their asso- The assembly of mitochondrial complex I involves building the ciation with 14 nuclear-encoded membrane subunits, the construc- 44 subunits emanating from two genomes into the two domains of tion of the extrinsic arm from 23 additional nuclear-encoded the complex. The enzyme is put together from preassembled sub- proteins, iron–sulfur clusters, and flavin mononucleotide cofactor complexes, and their subunit compositions have been characterized require the participation of assembly factors. Some are intrinsic to partially (14, 15). Extrinsic assembly factors of unknown function the complex, whereas others participate transiently. The suppres- become associated with subcomplexes that accumulate when as- sion of the expression of the NDUFA11 subunit of complex I dis- sembly and the activity of complex I are impaired by pathogenic rupted the assembly of the complex, and subcomplexes with mutations. Some assembly factor mutations also impair its activ- masses of 550 and 815 kDa accumulated. Eight of the known ex- ity (16). Other pathogenic mutations are found in all of the core trinsic assembly factors plus a hydrophobic protein, C3orf1, were subunits, and in 10 supernumerary subunits (NDUFA1, NDUFA2, associated with the subcomplexes. -

UNIVERSITY of CALIFORNIA, IRVINE Combinatorial Regulation By
UNIVERSITY OF CALIFORNIA, IRVINE Combinatorial regulation by maternal transcription factors during activation of the endoderm gene regulatory network DISSERTATION submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biological Sciences by Kitt D. Paraiso Dissertation Committee: Professor Ken W.Y. Cho, Chair Associate Professor Olivier Cinquin Professor Thomas Schilling 2018 Chapter 4 © 2017 Elsevier Ltd. © 2018 Kitt D. Paraiso DEDICATION To the incredibly intelligent and talented people, who in one way or another, helped complete this thesis. ii TABLE OF CONTENTS Page LIST OF FIGURES vii LIST OF TABLES ix LIST OF ABBREVIATIONS X ACKNOWLEDGEMENTS xi CURRICULUM VITAE xii ABSTRACT OF THE DISSERTATION xiv CHAPTER 1: Maternal transcription factors during early endoderm formation in 1 Xenopus Transcription factors co-regulate in a cell type-specific manner 2 Otx1 is expressed in a variety of cell lineages 4 Maternal otx1 in the endodermal conteXt 5 Establishment of enhancers by maternal transcription factors 9 Uncovering the endodermal gene regulatory network 12 Zygotic genome activation and temporal control of gene eXpression 14 The role of maternal transcription factors in early development 18 References 19 CHAPTER 2: Assembly of maternal transcription factors initiates the emergence 26 of tissue-specific zygotic cis-regulatory regions Introduction 28 Identification of maternal vegetally-localized transcription factors 31 Vegt and OtX1 combinatorially regulate the endodermal 33 transcriptome iii -

The Role of Cyclin B3 in Mammalian Meiosis
THE ROLE OF CYCLIN B3 IN MAMMALIAN MEIOSIS by Mehmet Erman Karasu A Dissertation Presented to the Faculty of the Louis V. Gerstner Jr. Graduate School of Biomedical Sciences, Memorial Sloan Kettering Cancer Center In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy New York, NY November, 2018 Scott Keeney, PhD Date Dissertation Mentor Copyright © Mehmet Erman Karasu 2018 DEDICATION I would like to dedicate this thesis to my parents, Mukaddes and Mustafa Karasu. I have been so lucky to have their support and unconditional love in this life. ii ABSTRACT Cyclins and cyclin dependent kinases (CDKs) lie at the center of the regulation of the cell cycle. Cyclins as regulatory partners of CDKs control the switch-like cell cycle transitions that orchestrate orderly duplication and segregation of genomes. Similar to somatic cell division, temporal regulation of cyclin-CDK activity is also important in meiosis, which is the specialized cell division that generates gametes for sexual production by halving the genome. Meiosis does so by carrying out one round of DNA replication followed by two successive divisions without another intervening phase of DNA replication. In budding yeast, cyclin-CDK activity has been shown to have a crucial role in meiotic events such as formation of meiotic double-strand breaks that initiate homologous recombination. Mammalian cells express numerous cyclins and CDKs, but how these proteins control meiosis remains poorly understood. Cyclin B3 was previously identified as germ cell specific, and its restricted expression pattern at the beginning of meiosis made it an interesting candidate to regulate meiotic events.