Whole-Exome Sequencing Identifies Recessive WDR62 Mutations in Severe Brain Malformations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genetical Society of Great Britain
Heredity 59 (1987) 151—160 The Genetical Society of Great Britain THEGENETICAL SOCIETY (Abstracts of papers presented at the TVVO HUNDRED AND FIFTH MEETING of the Society held on Friday, 14th and Saturday, 15th November 1986 at UNIVERSITY COLLEGE, LONDON) 1. Selection of somatic cell D. J. Porteous, P. A. Boyd, N. D. Hastie and hybrids with specific chromosome V. van Heyningen content for mapping the WAGR MAC Clinical and Population Cytogenetics Unit, Western General Hospital, Crewe Road, syndrome Edinburgh EH4 2XU. J. M. Fletcher, H. Morrison, J. A. Fantes, Clonedprobes for a number of available chromo- A. Seawright, S. Christie, D. J. Porteous, some ii assigned genes were used to define the N. D. Hastie and V. van Heyningen extent of deletions associated with the Wilms' MAC Clinical and Population Cytogenetics Unit, tumour, aniridia, genitourinary abnormalities and Western General Hospital, Crewe Road, mental retardation (WAGR) syndrome. Establish- Edinburgh EH4 2XU. ing reliable dosage studies for a number of different probes has proved difficult. We have therefore WAGR(Wilms tumour, aniridia, genitourinary abnormalities and mental retardation) syndrome concentrated on segregating the deleted chromo- is frequently associated with deletions on the short some 11 from a number of patients in somatic cell arm of chromosome 11. The deletions vary in size hybrids and analysing DNA from these to produce but always include part of band lipl3. To home a consistent map of chromosome lip. At the same in on the Wilms tumour and aniridia loci the end time we have determined the deletion breakpoints points of the different deletion breakpoints need at a molecular level and shown that the results are to be defined at the DNA level. -

Ciliopathiesneuromuscularciliopathies Disorders Disorders Ciliopathiesciliopathies
NeuromuscularCiliopathiesNeuromuscularCiliopathies Disorders Disorders CiliopathiesCiliopathies AboutAbout EGL EGL Genet Geneticsics EGLEGL Genetics Genetics specializes specializes in ingenetic genetic diagnostic diagnostic testing, testing, with with ne nearlyarly 50 50 years years of of clinical clinical experience experience and and board-certified board-certified labor laboratoryatory directorsdirectors and and genetic genetic counselors counselors reporting reporting out out cases. cases. EGL EGL Genet Geneticsics offers offers a combineda combined 1000 1000 molecular molecular genetics, genetics, biochemical biochemical genetics,genetics, and and cytogenetics cytogenetics tests tests under under one one roof roof and and custom custom test testinging for for all all medically medically relevant relevant genes, genes, for for domestic domestic andand international international clients. clients. EquallyEqually important important to to improving improving patient patient care care through through quality quality genetic genetic testing testing is is the the contribution contribution EGL EGL Genetics Genetics makes makes back back to to thethe scientific scientific and and medical medical communities. communities. EGL EGL Genetics Genetics is is one one of of only only a afew few clinical clinical diagnostic diagnostic laboratories laboratories to to openly openly share share data data withwith the the NCBI NCBI freely freely available available public public database database ClinVar ClinVar (>35,000 (>35,000 variants variants on on >1700 >1700 genes) genes) and and is isalso also the the only only laboratory laboratory with with a a frefree oen olinnlein dea dtabtaabsaes (eE m(EVmCVlaCslas)s,s f)e, afetuatruinrgin ag vaa vraiarniatn ctl acslasisfiscifiactiaotino sne saercahrc ahn adn rde rpeoprot rrte rqeuqeuset sint tinetrefarcfaec, ew, hwichhic fha cfailcitialiteatse rsa praidp id interactiveinteractive curation curation and and reporting reporting of of variants. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Ciliopathy-Associated Gene Cc2d2a Promotes Assembly of Subdistal Appendages on the Mother Centriole During Cilia Biogenesis
ARTICLE Received 7 Apr 2014 | Accepted 23 May 2014 | Published 20 Jun 2014 DOI: 10.1038/ncomms5207 Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis Shobi Veleri1, Souparnika H. Manjunath1, Robert N. Fariss2, Helen May-Simera1, Matthew Brooks1, Trevor A. Foskett1, Chun Gao2, Teresa A. Longo1, Pinghu Liu3, Kunio Nagashima4, Rivka A. Rachel1, Tiansen Li1, Lijin Dong3 & Anand Swaroop1 The primary cilium originates from the mother centriole and participates in critical functions during organogenesis. Defects in cilia biogenesis or function lead to pleiotropic phenotypes. Mutations in centrosome-cilia gene CC2D2A result in Meckel and Joubert syndromes. Here we generate a Cc2d2a À / À mouse that recapitulates features of Meckel syndrome including embryonic lethality and multiorgan defects. Cilia are absent in Cc2d2a À / À embryonic node and other somatic tissues; disruption of cilia-dependent Shh signalling appears to underlie exencephaly in mutant embryos. The Cc2d2a À / À mouse embryonic fibroblasts (MEFs) lack cilia, although mother centrioles and pericentriolar pro- teins are detected. Odf2, associated with subdistal appendages, is absent and ninein is reduced in mutant MEFs. In Cc2d2a À / À MEFs, subdistal appendages are lacking or abnormal by transmission electron microscopy. Consistent with this, CC2D2A localizes to subdistal appendages by immuno-EM in wild-type cells. We conclude that CC2D2A is essential for the assembly of subdistal appendages, which anchor cytoplasmic microtubules and prime the mother centriole for axoneme biogenesis. 1 Neurobiology-Neurodegeneration and Repair Laboratory, National Eye Institute, NIH, Bethesda, Maryland 20892, USA. 2 Biological Imaging Core, National Eye Institute, NIH, Bethesda, Maryland 20892, USA. -

Ciliopathies Gene Panel
Ciliopathies Gene Panel Contact details Introduction Regional Genetics Service The ciliopathies are a heterogeneous group of conditions with considerable phenotypic overlap. Levels 4-6, Barclay House These inherited diseases are caused by defects in cilia; hair-like projections present on most 37 Queen Square cells, with roles in key human developmental processes via their motility and signalling functions. Ciliopathies are often lethal and multiple organ systems are affected. Ciliopathies are London, WC1N 3BH united in being genetically heterogeneous conditions and the different subtypes can share T +44 (0) 20 7762 6888 many clinical features, predominantly cystic kidney disease, but also retinal, respiratory, F +44 (0) 20 7813 8578 skeletal, hepatic and neurological defects in addition to metabolic defects, laterality defects and polydactyly. Their clinical variability can make ciliopathies hard to recognise, reflecting the ubiquity of cilia. Gene panels currently offer the best solution to tackling analysis of genetically Samples required heterogeneous conditions such as the ciliopathies. Ciliopathies affect approximately 1:2,000 5ml venous blood in plastic EDTA births. bottles (>1ml from neonates) Ciliopathies are generally inherited in an autosomal recessive manner, with some autosomal Prenatal testing must be arranged dominant and X-linked exceptions. in advance, through a Clinical Genetics department if possible. Referrals Amniotic fluid or CV samples Patients presenting with a ciliopathy; due to the phenotypic variability this could be a diverse set should be sent to Cytogenetics for of features. For guidance contact the laboratory or Dr Hannah Mitchison dissecting and culturing, with ([email protected]) / Prof Phil Beales ([email protected]) instructions to forward the sample to the Regional Molecular Genetics Referrals will be accepted from clinical geneticists and consultants in nephrology, metabolic, laboratory for analysis respiratory and retinal diseases. -

Identification and Characterization of Genes Essential for Human Brain Development
Identification and Characterization of Genes Essential for Human Brain Development The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Ganesh, Vijay S. 2012. Identification and Characterization of Genes Essential for Human Brain Development. Doctoral dissertation, Harvard University. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:9773743 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Copyright © 2012 by Vijay S. Ganesh All rights reserved. Dissertation Advisor: Dr. Christopher A. Walsh Author: Vijay S. Ganesh Identification and Characterization of Genes Essential for Human Brain Development Abstract The human brain is a network of ninety billion neurons that allows for many of the behavioral adaptations considered unique to our species. One-fifth of these neurons are layered in an epithelial sheet known as the cerebral cortex, which is exquisitely folded into convolutions called gyri. Defects in neuronal number clinically present with microcephaly (Greek for “small head”), and in inherited cases these defects can be linked to mutations that identify genes essential for neural progenitor proliferation. Most microcephaly genes are characterized to play a role in the centrosome, however rarer presentations of microcephaly have identified different mechanisms. Charged multivesicular body protein/Chromatin modifying protein 1A (CHMP1A) is a member of the ESCRT-III endosomal sorting complex, but is also suggested to localize to the nuclear matrix and regulate chromatin. -

Molecular Genetics of Microcephaly Primary Hereditary: an Overview
brain sciences Review Molecular Genetics of Microcephaly Primary Hereditary: An Overview Nikistratos Siskos † , Electra Stylianopoulou †, Georgios Skavdis and Maria E. Grigoriou * Department of Molecular Biology & Genetics, Democritus University of Thrace, 68100 Alexandroupolis, Greece; [email protected] (N.S.); [email protected] (E.S.); [email protected] (G.S.) * Correspondence: [email protected] † Equal contribution. Abstract: MicroCephaly Primary Hereditary (MCPH) is a rare congenital neurodevelopmental disorder characterized by a significant reduction of the occipitofrontal head circumference and mild to moderate mental disability. Patients have small brains, though with overall normal architecture; therefore, studying MCPH can reveal not only the pathological mechanisms leading to this condition, but also the mechanisms operating during normal development. MCPH is genetically heterogeneous, with 27 genes listed so far in the Online Mendelian Inheritance in Man (OMIM) database. In this review, we discuss the role of MCPH proteins and delineate the molecular mechanisms and common pathways in which they participate. Keywords: microcephaly; MCPH; MCPH1–MCPH27; molecular genetics; cell cycle 1. Introduction Citation: Siskos, N.; Stylianopoulou, Microcephaly, from the Greek word µικρoκεϕαλi´α (mikrokephalia), meaning small E.; Skavdis, G.; Grigoriou, M.E. head, is a term used to describe a cranium with reduction of the occipitofrontal head circum- Molecular Genetics of Microcephaly ference equal, or more that teo standard deviations -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Chromosome 6 Abnormalities in Ovarian Surface Epithelial Tumors of Borderline Malignancy Suggest a Genetic Continuum in the Progression Model of Ovarian Neoplasms1
3404 Vol. 7, 3404–3409, November 2001 Clinical Cancer Research Chromosome 6 Abnormalities in Ovarian Surface Epithelial Tumors of Borderline Malignancy Suggest a Genetic Continuum in the Progression Model of Ovarian Neoplasms1 Maria Grazia Tibiletti, Barbara Bernasconi, Conclusion: Alterations in the above-mentioned inter- Daniela Furlan, Paola Bressan, Roberta Cerutti, val are a common finding in advanced ovarian carcinomas but also in benign ovarian cysts, implying that some tumors Carla Facco, Massimo Franchi, Cristina Riva, of borderline malignancy may arise from benign tumors and Raffaella Cinquetti, Carlo Capella, and that malignant ones may evolve from tumors of borderline Roberto Taramelli2 malignancy. Genes located in 6q27 seem to be crucial for this Laboratorio di Anatomia Patologica Ospedale di Circolo, Fondazione mechanism of early events in ovarian tumorigenesis. Macchi, 21100 Varese [M. G. T., C. F., C. R.]; and Dipartimento di Scienze Cliniche e Biologiche [B. B., D. F., P. B., R. Ce., C. C.], INTRODUCTION Clinica Ostetrica e Ginecologica [M. F.], and Dipartimento di Biologia Strutturale e Funzionale [R. Ci., R. T.], Universita` Ovarian cancer represents the most lethal of the gyneco- dell’Insubria, 21100 Varese, Italy logical tumors, and its pathogenesis is poorly understood. More than 80% of malignant tumors of the ovary are of epithelial origin and arise from the surface epithelium (1). Ovarian surface ABSTRACT epithelial tumors are divided into three histological subtypes: Purpose: We used conventional cytogenetics, molecular benign cystoadenoma, TBM,3 and AC, which are related to a cytogenetics, and molecular genetic analyses to study the different biological behavior (1). The molecular events under- pattern of allelic loss on chromosome 6q in a cohort of lying the development of ovarian tumorigenesis are still largely borderline epithelial ovarian tumors. -
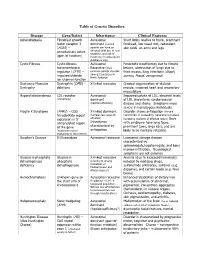
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical Features Achondroplasia Fibroblast growth Autosomal Short limbs relative to trunk, prominent factor receptor 3 dominant (normal forehead, low nasal root, redundant (FGR3) – parents can have an skin folds on arms and legs constitutively active affected child due to new mutation, and risk of (gain of function) recurrence in subsequent children is low) Cystic Fibrosis Cystic fibrosis Autosomal Pancreatic insufficiency due to fibrotic transmembrane Recessive (most lesions, obstruction of lungs due to regulator (CFTR) – common genetic disorder thick mucus, lung infections (Staph, impaired chloride among Caucasians in aureus, Pseud. aeruginosa) North America) ion channel function Duchenne Muscular Dystrophin (DMD) - X-linked recessive Gradual degeneration of skeletal Dystrophy deletions muscle, impaired heart and respiratory musculature Hypercholesterolemia LDL receptor Autosomal Impaired uptake of LDL, elevated levels (commonly) dominant of LDL cholesterol, cardiovascular (haploinsufficiency) disease and stroke. Symptoms more severe in homozygous individuals Fragile X Syndrome (FMR1) – CGG X-linked dominant Disorder shows anticipation (female trinucleotide repeat (females less severely transmitters in succeeding generations produce expansion in 5’ affected) increasing numbers of affected males) Boys untranslated region Inheritance with syndrome have long faces, of the gene characterized by prominent jaws, large ears, and are (expansion occurs anticipation likely to be mentally retarded. exclusively in the mother) Gaucher’s Disease Β-Glucosidase Autosomal recessive Lysosomal storage disease characterized by splenomegaly,hepatomegaly, and bone marrow infiltration. Neurological symptoms are not common Glucose 6-phosphate Glucose 6- X-linked recessive Anemia (due to increased hemolysis) dehydrogenase phosphate (prominent among induced by oxidizing drugs, deficiency dehydrogenase individuals of sulfonamide antibiotics, sulfones (e.g. -

Ncomms4885.Pdf
ARTICLE Received 4 Feb 2014 | Accepted 14 Apr 2014 | Published 30 May 2014 DOI: 10.1038/ncomms4885 Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size Jian-Fu Chen1,2, Ying Zhang1, Jonathan Wilde1, Kirk C. Hansen3, Fan Lai4 & Lee Niswander1 Human genetic studies have established a link between a class of centrosome proteins and microcephaly. Current studies of microcephaly focus on defective centrosome/spindle orientation. Mutations in WDR62 are associated with microcephaly and other cortical abnormalities in humans. Here we create a mouse model of Wdr62 deficiency and find that the mice exhibit reduced brain size due to decreased neural progenitor cells (NPCs). Wdr62 depleted cells show spindle instability, spindle assembly checkpoint (SAC) activation, mitotic arrest and cell death. Mechanistically, Wdr62 associates and genetically interacts with Aurora A to regulate spindle formation, mitotic progression and brain size. Our results suggest that Wdr62 interacts with Aurora A to control mitotic progression, and loss of these interactions leads to mitotic delay and cell death of NPCs, which could be a potential cause of human microcephaly. 1 Howard Hughes Medical Institute, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Children’s Hospital Colorado, Aurora, Colorado 80045, USA. 2 Department of Genetics, Department of Biochemistry & Molecular Biology, University of Georgia, Athens, Georgia 30602, USA. 3 Biochemistry & Molecular Genetics, University of Colorado Denver, Aurora, Colorado 80045, USA. 4 The Wistar Institute, 3601 Spruce Street, Philadelphia, Pennsylvania 19104, USA. Correspondence and requests for materials should be addressed to J-F.C. (email: [email protected]) or to L.N. -

Molecular Cytogenetic Characterisation of a Novel De Novo Ring
Pace et al. Molecular Cytogenetics (2017) 10:9 DOI 10.1186/s13039-017-0311-y CASEREPORT Open Access Molecular cytogenetic characterisation of a novel de novo ring chromosome 6 involving a terminal 6p deletion and terminal 6q duplication in the different arms of the same chromosome Nikolai Paul Pace1, Frideriki Maggouta2, Melissa Twigden2 and Isabella Borg1,3,4* Abstract Background: Ring chromosome 6 is a rare sporadic chromosomal abnormality, associated with extreme variability in clinical phenotypes. Most ring chromosomes are known to have deletions on one or both chromosomal arms. Here, we report an atypical and unique ring chromosome 6 involving both a distal deletion and a distal duplication on the different arms of the same chromosome. Case presentation: In a patient with intellectual disability, short stature, microcephaly, facial dysmorphology, congenital heart defects and renovascular disease, a ring chromosome 6 was characterised using array-CGH and dual-colour FISH. The de-novo ring chromosome 6 involved a 1.8 Mb terminal deletion in the distal short arm and a 2.5 Mb duplication in the distal long arm of the same chromosome 6. This results in monosomy for the region 6pter to 6p25.3 and trisomy for the region 6q27 to 6qter. Analysis of genes in these chromosomal regions suggests that haploinsufficiency for FOXC1 and GMDS genes accounts for the cardiac and neurodevelopmental phenotypes in the proband. The ring chromosome 6 reported here is atypical as it involves a unique duplication of the distal long arm. Furthermore, the presence of renovascular disease is also a unique feature identified in this patient.