6Q Deletions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genetical Society of Great Britain
Heredity 59 (1987) 151—160 The Genetical Society of Great Britain THEGENETICAL SOCIETY (Abstracts of papers presented at the TVVO HUNDRED AND FIFTH MEETING of the Society held on Friday, 14th and Saturday, 15th November 1986 at UNIVERSITY COLLEGE, LONDON) 1. Selection of somatic cell D. J. Porteous, P. A. Boyd, N. D. Hastie and hybrids with specific chromosome V. van Heyningen content for mapping the WAGR MAC Clinical and Population Cytogenetics Unit, Western General Hospital, Crewe Road, syndrome Edinburgh EH4 2XU. J. M. Fletcher, H. Morrison, J. A. Fantes, Clonedprobes for a number of available chromo- A. Seawright, S. Christie, D. J. Porteous, some ii assigned genes were used to define the N. D. Hastie and V. van Heyningen extent of deletions associated with the Wilms' MAC Clinical and Population Cytogenetics Unit, tumour, aniridia, genitourinary abnormalities and Western General Hospital, Crewe Road, mental retardation (WAGR) syndrome. Establish- Edinburgh EH4 2XU. ing reliable dosage studies for a number of different probes has proved difficult. We have therefore WAGR(Wilms tumour, aniridia, genitourinary abnormalities and mental retardation) syndrome concentrated on segregating the deleted chromo- is frequently associated with deletions on the short some 11 from a number of patients in somatic cell arm of chromosome 11. The deletions vary in size hybrids and analysing DNA from these to produce but always include part of band lipl3. To home a consistent map of chromosome lip. At the same in on the Wilms tumour and aniridia loci the end time we have determined the deletion breakpoints points of the different deletion breakpoints need at a molecular level and shown that the results are to be defined at the DNA level. -

Pierre Robin and the Syndrome That Bears His Name PETER RANDALL
Pierre Robin and the Syndrome That Bears His Name PETER RANDALL, M.D. WILTON M. KROGMAN, Ph.D. SOONA JAHINA, B.D.S., M.Sc. Philadelphia, Pennsylvania The Pierre Robin Syndrome refers to a combination of micrognathia (a small jaw) and glossoptosis (literally, a falling downward or back- ward of the tongue) in the newborn infant (Figure 1). These conditions are likely to cause obstruction of the upper airway, and they are fre- quently associated with an incomplete cleft of the palate. Patients with the Pierre Robin Syndrome may present a real emer- gency in the delivery room because of the obstructed upper airway, or the airway problem may not become manifest for several days or weeks (10, 11, 38). There is frequently a feeding problem, as well as problems associated with the cleft of the palate (if one is present) and also an unusual malocclusion (2, 5, 12, 16). In addition, it presents a fascinating anthropological puzzle (22, 23). This paper will review the work of Dr. Robin, consider some possible etiologies of this syndrome, and report on some work on mandibular bone growth in a group of such patients. History Pierre Robin was far from the first person to recognize this syndrome. One account is recorded in 1822 by St. Hilaire. In 1891 Taruffi men- tioned two subclassifications-hypomicrognatus (small jaw) and hypo- agnathus (absent jaw). In 1891, four cases, two of them having cleft palates, were reported by Lanneloague and Monard (12, 14). Shukow- sky in 1902 described a tongue to lip surgical adhesion to overcome the respiratory obstruction (34). -

Podo Pediatrics Podo Pediatrics
Podo Pediatrics Identifying Biomechanical Pathologies David Lee, D.P.M., D. A.B.P.S. Purpose • Identification of mechanical foot and ankle conditions • Base treatments • Knowing when to refer to a podiatrist Topics • Flatfoot (Pes Plano Valgus) • Equinus • Intoed feet (Cavo-adductor Varus) • Heel pain (Calcaneodynia) • Shin Splints • Various Pedal deformities 1 WHAT IS NORMAL? At birth to ~9 months • Ankle flexible to over 20 deg DF • No “C” shaped foot • No clicking or popping sounds • Babinski sign • Pull up 7-8mo. 9-16 months… • Begin walking • Feet are fat, flat and floppy • Knees are always center or externally rotated, never internal. • Stance is wide and less stable • Stomping gait pattern 2 16-18 months • Able to walk upstairs • Knee never internal • Still wide base and flat and floppy feet • Stomping still 3-7 years • Able toe walk downstairs • Heel-to-toe walk • Watch for – Intoeing – Tripping – Tight ankle joint (equinus) 7 years and up • Arch should be developed • Heel-to-toe walk • Heel is perpendicular to ground • Knees straight ahead 3 Neutral Internal Rotation Early detection is important • Prevent long term adaptation • Joint damage • Adult pathology – Heel pain, bunions, hammertoes, ankle instability, knee pain, shin splints, etc. • Ability to thrive physically and socially 4 THE FLAT FOOT Visual Complaints by the Parent • Tripping or falling • Poor balance- Clumsy • Feet look funny, walks funny • Shoes wearing out quickly Social Complaints by the Parent • Lazy, inactive, “doesn’t like going outside to play or play sports -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Chromosome 6 Abnormalities in Ovarian Surface Epithelial Tumors of Borderline Malignancy Suggest a Genetic Continuum in the Progression Model of Ovarian Neoplasms1
3404 Vol. 7, 3404–3409, November 2001 Clinical Cancer Research Chromosome 6 Abnormalities in Ovarian Surface Epithelial Tumors of Borderline Malignancy Suggest a Genetic Continuum in the Progression Model of Ovarian Neoplasms1 Maria Grazia Tibiletti, Barbara Bernasconi, Conclusion: Alterations in the above-mentioned inter- Daniela Furlan, Paola Bressan, Roberta Cerutti, val are a common finding in advanced ovarian carcinomas but also in benign ovarian cysts, implying that some tumors Carla Facco, Massimo Franchi, Cristina Riva, of borderline malignancy may arise from benign tumors and Raffaella Cinquetti, Carlo Capella, and that malignant ones may evolve from tumors of borderline Roberto Taramelli2 malignancy. Genes located in 6q27 seem to be crucial for this Laboratorio di Anatomia Patologica Ospedale di Circolo, Fondazione mechanism of early events in ovarian tumorigenesis. Macchi, 21100 Varese [M. G. T., C. F., C. R.]; and Dipartimento di Scienze Cliniche e Biologiche [B. B., D. F., P. B., R. Ce., C. C.], INTRODUCTION Clinica Ostetrica e Ginecologica [M. F.], and Dipartimento di Biologia Strutturale e Funzionale [R. Ci., R. T.], Universita` Ovarian cancer represents the most lethal of the gyneco- dell’Insubria, 21100 Varese, Italy logical tumors, and its pathogenesis is poorly understood. More than 80% of malignant tumors of the ovary are of epithelial origin and arise from the surface epithelium (1). Ovarian surface ABSTRACT epithelial tumors are divided into three histological subtypes: Purpose: We used conventional cytogenetics, molecular benign cystoadenoma, TBM,3 and AC, which are related to a cytogenetics, and molecular genetic analyses to study the different biological behavior (1). The molecular events under- pattern of allelic loss on chromosome 6q in a cohort of lying the development of ovarian tumorigenesis are still largely borderline epithelial ovarian tumors. -

Flexible Flatfoot
REVIEW ORTHOPEDICS & TRAUMATOLOGY North Clin Istanbul 2014;1(1):57-64 doi: 10.14744/nci.2014.29292 Flexible flatfoot Aziz Atik1, Selahattin Ozyurek2 1Department of Orthopedics and Tarumatology, Balikesir University Faculty of Medicine, Balikesir, Turkey; 2Department of Orthopedics and Traumatology, Aksaz Military Hospital, Marmaris, Mugla, Turkey ABSTRACT While being one of the most frequent parental complained deformities, flatfoot does not have a universally ac- cepted description. The reasons of flexible flatfoot are still on debate, but they must be differentiated from rigid flatfoot which occurs secondary to other pathologies. These children are commonly brought up to a physician without any complaint. It should be kept in mind that the etiology may vary from general soft tissue laxities to intrinsic foot pathologies. Every flexible flatfoot does not require radiological examination or treatment if there is no complaint. Otherwise further investigation and conservative or surgical treatment may necessitate. Key words: Children; flatfoot; flexible; foot problem; pes planus. hough the term flatfoot (pes planus) is gener- forms again (Figure 2). When weight-bearing forces Tally defined as a condition which the longitu- on feet are relieved this arch can be observed. If the dinal arch of the foot collapses, it has not a clinically foot is not bearing any weight, still medial longitu- or radiologically accepted universal definition. Flat- dinal arch is not seen, then it is called rigid (fixed) foot which we frequently encounter in routine out- flatfoot. To differentiate between these two condi- patient practice will be more accurately seen as a re- tions easily, Jack’s test (great toe is dorisflexed as the sult of laxity of ligaments of the foot. -
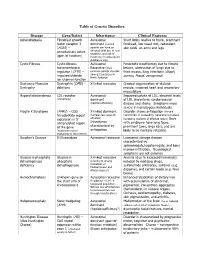
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical Features Achondroplasia Fibroblast growth Autosomal Short limbs relative to trunk, prominent factor receptor 3 dominant (normal forehead, low nasal root, redundant (FGR3) – parents can have an skin folds on arms and legs constitutively active affected child due to new mutation, and risk of (gain of function) recurrence in subsequent children is low) Cystic Fibrosis Cystic fibrosis Autosomal Pancreatic insufficiency due to fibrotic transmembrane Recessive (most lesions, obstruction of lungs due to regulator (CFTR) – common genetic disorder thick mucus, lung infections (Staph, impaired chloride among Caucasians in aureus, Pseud. aeruginosa) North America) ion channel function Duchenne Muscular Dystrophin (DMD) - X-linked recessive Gradual degeneration of skeletal Dystrophy deletions muscle, impaired heart and respiratory musculature Hypercholesterolemia LDL receptor Autosomal Impaired uptake of LDL, elevated levels (commonly) dominant of LDL cholesterol, cardiovascular (haploinsufficiency) disease and stroke. Symptoms more severe in homozygous individuals Fragile X Syndrome (FMR1) – CGG X-linked dominant Disorder shows anticipation (female trinucleotide repeat (females less severely transmitters in succeeding generations produce expansion in 5’ affected) increasing numbers of affected males) Boys untranslated region Inheritance with syndrome have long faces, of the gene characterized by prominent jaws, large ears, and are (expansion occurs anticipation likely to be mentally retarded. exclusively in the mother) Gaucher’s Disease Β-Glucosidase Autosomal recessive Lysosomal storage disease characterized by splenomegaly,hepatomegaly, and bone marrow infiltration. Neurological symptoms are not common Glucose 6-phosphate Glucose 6- X-linked recessive Anemia (due to increased hemolysis) dehydrogenase phosphate (prominent among induced by oxidizing drugs, deficiency dehydrogenase individuals of sulfonamide antibiotics, sulfones (e.g. -

Molecular Cytogenetic Characterisation of a Novel De Novo Ring
Pace et al. Molecular Cytogenetics (2017) 10:9 DOI 10.1186/s13039-017-0311-y CASEREPORT Open Access Molecular cytogenetic characterisation of a novel de novo ring chromosome 6 involving a terminal 6p deletion and terminal 6q duplication in the different arms of the same chromosome Nikolai Paul Pace1, Frideriki Maggouta2, Melissa Twigden2 and Isabella Borg1,3,4* Abstract Background: Ring chromosome 6 is a rare sporadic chromosomal abnormality, associated with extreme variability in clinical phenotypes. Most ring chromosomes are known to have deletions on one or both chromosomal arms. Here, we report an atypical and unique ring chromosome 6 involving both a distal deletion and a distal duplication on the different arms of the same chromosome. Case presentation: In a patient with intellectual disability, short stature, microcephaly, facial dysmorphology, congenital heart defects and renovascular disease, a ring chromosome 6 was characterised using array-CGH and dual-colour FISH. The de-novo ring chromosome 6 involved a 1.8 Mb terminal deletion in the distal short arm and a 2.5 Mb duplication in the distal long arm of the same chromosome 6. This results in monosomy for the region 6pter to 6p25.3 and trisomy for the region 6q27 to 6qter. Analysis of genes in these chromosomal regions suggests that haploinsufficiency for FOXC1 and GMDS genes accounts for the cardiac and neurodevelopmental phenotypes in the proband. The ring chromosome 6 reported here is atypical as it involves a unique duplication of the distal long arm. Furthermore, the presence of renovascular disease is also a unique feature identified in this patient. -

WNT16 Is a New Marker of Senescence
Table S1. A. Complete list of 177 genes overexpressed in replicative senescence Value Gene Description UniGene RefSeq 2.440 WNT16 wingless-type MMTV integration site family, member 16 (WNT16), transcript variant 2, mRNA. Hs.272375 NM_016087 2.355 MMP10 matrix metallopeptidase 10 (stromelysin 2) (MMP10), mRNA. Hs.2258 NM_002425 2.344 MMP3 matrix metallopeptidase 3 (stromelysin 1, progelatinase) (MMP3), mRNA. Hs.375129 NM_002422 2.300 HIST1H2AC Histone cluster 1, H2ac Hs.484950 2.134 CLDN1 claudin 1 (CLDN1), mRNA. Hs.439060 NM_021101 2.119 TSPAN13 tetraspanin 13 (TSPAN13), mRNA. Hs.364544 NM_014399 2.112 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 2.070 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 2.026 DCBLD2 discoidin, CUB and LCCL domain containing 2 (DCBLD2), mRNA. Hs.203691 NM_080927 2.007 SERPINB2 serpin peptidase inhibitor, clade B (ovalbumin), member 2 (SERPINB2), mRNA. Hs.594481 NM_002575 2.004 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.989 OBFC2A Oligonucleotide/oligosaccharide-binding fold containing 2A Hs.591610 1.962 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.947 PLCB4 phospholipase C, beta 4 (PLCB4), transcript variant 2, mRNA. Hs.472101 NM_182797 1.934 PLCB4 phospholipase C, beta 4 (PLCB4), transcript variant 1, mRNA. Hs.472101 NM_000933 1.933 KRTAP1-5 keratin associated protein 1-5 (KRTAP1-5), mRNA. Hs.534499 NM_031957 1.894 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.884 CYTL1 cytokine-like 1 (CYTL1), mRNA. Hs.13872 NM_018659 tumor necrosis factor receptor superfamily, member 10d, decoy with truncated death domain (TNFRSF10D), 1.848 TNFRSF10D Hs.213467 NM_003840 mRNA. -

Isolation of Chromosome-Specific Ests by Microdissection-Mediated Cdna Capture Edgardo Gracia, 1-3 Michael E
Downloaded from genome.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press RESEARCH Isolation of Chromosome-Specific ESTs by Microdissection-Mediated cDNA Capture Edgardo Gracia, 1-3 Michael E. Ray, 1-3 Mihael H. Polymeropoulos, 4 Anindya Dehejia, 4 Paul S. Meltzer, 3 and Jeffrey M. Trent 3's 1Department of Human Genetics, The University of Michigan Medical School, Medical Science II M4708, Ann Arbor, Michigan 48109; 3Laboratory of Cancer Genetics, 4 Laboratory of Genetic Disease Research, National Center for Human Genome Research, National Institutes of Health, Bethesda, Maryland 20892 Despite dramatic advances in the identification of human expressed sequence tags (ESTs), techniques that facilitate isolation of chromosome or chromosome band-specific ESTs would be of considerable value. This report demonstrates the feasibility of identifying chromosome-specific ESTs following microdissection of a single-copy chromosome region. For this study, a reduced complexity cDNA library was linkered and hybridized to normal human metaphase chromosomes. After stringency washes, the entire long arm of chromosome 6 (6q) was microdissected. Following PCR amplification using linker-specific primers, captured cDNAs were subcloned and 187 individual clones picked at random. These 187 clones were then sorted by filter cross-hybridization into 34 unique groups. Of these 34 groups, 19 (56%) mapped to chromosome 6 by Southern blot. We identified three previously known genes, human cytovillin (ezrin) mapped previously to 6q25-26, human cardiac gap junction protein (connexin 43) mapped previously to 6q21-23.2 and prolyloligopeptidase, which had not been mapped previously. BLASTN identified three clone groups with homology to known ESTs and 12 representing novel cDNA sequences. -

Foot Function Disorders in Children with Severe Spondylolisthesis of L5 Vertebra I.E
СLINICAL STUDIES УДК 617.586-053.2:616.721.7-001.7 DOI: 10.21823/2311-2905-2019-25-2-71-80 Foot Function Disorders in Children with Severe Spondylolisthesis of L5 Vertebra I.E. Nikityuk, S.V. Vissarionov Turner Scientific Research Institute for Children’s Orthopedics, St. Petersburg, Russian Federation Abstract Background. In children with spondylolisthesis, there are still unexplained aspects in the relationship of the degree of L5 displacement with the severity of the clinical picture and neurological disorders. At the same time, aspects of the mutual aggravating influence of the indicated spinal disorder on the condition of the feet have not been studied. Therefore, the problem of identifying disorder of foot function in children with spinal L5 spondylolisthesis is relevant. Aim of the study — to evaluate the deviations in parameters of the transverse and longitudinal arches of feet in children with severe L5 spondylolisthesis. Materials and Methods. In the period from 2016 to 2018, 12 children aged 14.1 y.o. [12,7; 15,5] were examined with L5 spondylolisthesis body of grade III–IV, accompanied by stenosis of the spinal canal at the same level and by compression of the roots of the spinal cord. Imaging diagnostics included multispiral computed tomography (MSCT) and magnetic resonance imaging (MRI). To estimate the function of the feet, double-bearing and single-bearing plantography was used. The data for the control group included only plantographic examinations of 12 healthy children of the same age. Results. In patients with spondylolisthesis, the mean value of the anterior t and intermediate s plantographic bearing indices were significantly lower than those of healthy children. -

Of Small Intestine Harboring Driver Gene Mutations: a Case Report and a Literature Review
1161 Case Report A rare multiple primary sarcomatoid carcinoma (SCA) of small intestine harboring driver gene mutations: a case report and a literature review Zhu Zhu1#, Xinyi Liu2#, Wenliang Li1, Zhengqi Wen1, Xiang Ji1, Ruize Zhou1, Xiaoyu Tuo3, Yaru Chen2, Xian Gong2, Guifeng Liu2, Yanqing Zhou2, Shifu Chen2, Lele Song2#^, Jian Huang1 1Department of Oncology, First Affiliated Hospital of Kunming Medical University, Kunming, China; 2HaploX Biotechnology, Shenzhen, China; 3Department of Pathology, First Affiliated Hospital of Kunming Medical University, Kunming, China #These authors contributed equally to this work. Correspondence to: Jian Huang. Department of Oncology, First Affiliated Hospital of Kunming Medical University, No. 295, Xichang Road, Kunming 560032, Yunnan Province, China. Email: [email protected]; Lele Song. HaploX Biotechnology, 8th floor, Auto Electric Power Building, Songpingshan Road, Nanshan District, Shenzhen 518057, Guangdong Province, China. Email: [email protected]. Abstract: Primary sarcomatoid carcinoma (SCA) is a type of rare tumor consisting of both malignant epithelial and mesenchymal components. Only 32 cases of SCA of the small bowel have been reported in the literature to date. Due to its rarity and complexity, this cancer has not been genetically studied and its diagnosis and treatment remain difficult. Here we report a 54-year-old male underwent emergency surgical resection in the small intestine due to severe obstruction and was diagnosed with multiple SCA based on postoperative pathological examination. Over 100 polypoid tumors scattered along his whole jejunum and proximal ileum. Chemotherapy (IFO+Epirubicin) was performed after surgery while the patient died two months after the surgery due to severe malnutrition. Whole-exome sequencing was performed for the tumor tissue with normal tissue as the control.