Bones and Joints in Marfan Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Challenges in Long-Term Control of Hypercalcaemia with Denosumab
Taylor-Miller, T., Sivaprakasam, P., Smithson, S. F., Steward, C. G., & Burren, C. P. (2021). Challenges in long-term control of hypercalcaemia with denosumab after haematopoietic stem cell transplantation for TNFRSF11A osteoclast-poor autosomal recessive osteopetrosis. Bone reports, 14, 100738. [100738]. https://doi.org/10.1016/j.bonr.2020.100738 Publisher's PDF, also known as Version of record License (if available): CC BY-NC-ND Link to published version (if available): 10.1016/j.bonr.2020.100738 Link to publication record in Explore Bristol Research PDF-document This is the final published version of the article (version of record). It first appeared online via Elsevier at https://doi.org/10.1016/j.bonr.2020.100738. Please refer to any applicable terms of use of the publisher. University of Bristol - Explore Bristol Research General rights This document is made available in accordance with publisher policies. Please cite only the published version using the reference above. Full terms of use are available: http://www.bristol.ac.uk/red/research-policy/pure/user-guides/ebr-terms/ Bone Reports 14 (2021) 100738 Contents lists available at ScienceDirect Bone Reports journal homepage: www.elsevier.com/locate/bonr Case Report Challenges in long-term control of hypercalcaemia with denosumab after haematopoietic stem cell transplantation for TNFRSF11A osteoclast-poor autosomal recessive osteopetrosis Tashunka Taylor-Miller a, Ponni Sivaprakasam b, Sarah F. Smithson c,d, Colin G. Steward d,e, Christine P. Burren a,d,* a Department of Paediatric -

Pierre Robin and the Syndrome That Bears His Name PETER RANDALL
Pierre Robin and the Syndrome That Bears His Name PETER RANDALL, M.D. WILTON M. KROGMAN, Ph.D. SOONA JAHINA, B.D.S., M.Sc. Philadelphia, Pennsylvania The Pierre Robin Syndrome refers to a combination of micrognathia (a small jaw) and glossoptosis (literally, a falling downward or back- ward of the tongue) in the newborn infant (Figure 1). These conditions are likely to cause obstruction of the upper airway, and they are fre- quently associated with an incomplete cleft of the palate. Patients with the Pierre Robin Syndrome may present a real emer- gency in the delivery room because of the obstructed upper airway, or the airway problem may not become manifest for several days or weeks (10, 11, 38). There is frequently a feeding problem, as well as problems associated with the cleft of the palate (if one is present) and also an unusual malocclusion (2, 5, 12, 16). In addition, it presents a fascinating anthropological puzzle (22, 23). This paper will review the work of Dr. Robin, consider some possible etiologies of this syndrome, and report on some work on mandibular bone growth in a group of such patients. History Pierre Robin was far from the first person to recognize this syndrome. One account is recorded in 1822 by St. Hilaire. In 1891 Taruffi men- tioned two subclassifications-hypomicrognatus (small jaw) and hypo- agnathus (absent jaw). In 1891, four cases, two of them having cleft palates, were reported by Lanneloague and Monard (12, 14). Shukow- sky in 1902 described a tongue to lip surgical adhesion to overcome the respiratory obstruction (34). -
Spinal Deformity Study Group
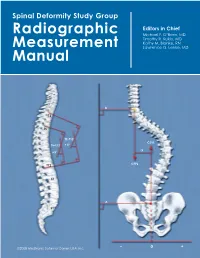
Spinal Deformity Study Group Editors in Chief Radiographic Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Measurement Lawrence G. Lenke, MD Manual B T2 T5 T2–T12 CSVL T5–T12 +X° -X +X° C7PL T12 L2 A S1 ©2008 Medtronic Sofamor Danek USA, Inc. – 0 + Radiographic Measurement Manual Editors in Chief Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Lawrence G. Lenke, MD Section Editors Keith H. Bridwell, MD Kathy M. Blanke, RN Christopher L. Hamill, MD William C. Horton, MD Timothy R. Kuklo, MD Hubert B. Labelle, MD Lawrence G. Lenke, MD Michael F. O’Brien, MD David W. Polly Jr, MD B. Stephens Richards III, MD Pierre Roussouly, MD James O. Sanders, MD ©2008 Medtronic Sofamor Danek USA, Inc. Acknowledgements Radiographic Measurement Manual The radiographic measurement manual has been developed to present standardized techniques for radiographic measurement. In addition, this manual will serve as a complimentary guide for the Spinal Deformity Study Group’s radiographic measurement software. Special thanks to the following members of the Spinal Deformity Study Group in the development of this manual. Sigurd Berven, MD Hubert B. Labelle, MD Randal Betz, MD Lawrence G. Lenke, MD Fabien D. Bitan, MD Thomas G. Lowe, MD John T. Braun, MD John P. Lubicky, MD Keith H. Bridwell, MD Steven M. Mardjetko, MD Courtney W. Brown, MD Richard E. McCarthy, MD Daniel H. Chopin, MD Andrew A. Merola, MD Edgar G. Dawson, MD Michael Neuwirth, MD Christopher DeWald, MD Peter O. Newton, MD Mohammad Diab, MD Michael F. -

Podo Pediatrics Podo Pediatrics
Podo Pediatrics Identifying Biomechanical Pathologies David Lee, D.P.M., D. A.B.P.S. Purpose • Identification of mechanical foot and ankle conditions • Base treatments • Knowing when to refer to a podiatrist Topics • Flatfoot (Pes Plano Valgus) • Equinus • Intoed feet (Cavo-adductor Varus) • Heel pain (Calcaneodynia) • Shin Splints • Various Pedal deformities 1 WHAT IS NORMAL? At birth to ~9 months • Ankle flexible to over 20 deg DF • No “C” shaped foot • No clicking or popping sounds • Babinski sign • Pull up 7-8mo. 9-16 months… • Begin walking • Feet are fat, flat and floppy • Knees are always center or externally rotated, never internal. • Stance is wide and less stable • Stomping gait pattern 2 16-18 months • Able to walk upstairs • Knee never internal • Still wide base and flat and floppy feet • Stomping still 3-7 years • Able toe walk downstairs • Heel-to-toe walk • Watch for – Intoeing – Tripping – Tight ankle joint (equinus) 7 years and up • Arch should be developed • Heel-to-toe walk • Heel is perpendicular to ground • Knees straight ahead 3 Neutral Internal Rotation Early detection is important • Prevent long term adaptation • Joint damage • Adult pathology – Heel pain, bunions, hammertoes, ankle instability, knee pain, shin splints, etc. • Ability to thrive physically and socially 4 THE FLAT FOOT Visual Complaints by the Parent • Tripping or falling • Poor balance- Clumsy • Feet look funny, walks funny • Shoes wearing out quickly Social Complaints by the Parent • Lazy, inactive, “doesn’t like going outside to play or play sports -

Lordosis, Kyphosis, and Scoliosis
SPINAL CURVATURES: LORDOSIS, KYPHOSIS, AND SCOLIOSIS The human spine normally curves to aid in stability or balance and to assist in absorbing shock during movement. These gentle curves can be seen from the side or lateral view of the spine. When viewed from the back, the spine should run straight down the middle of the back. When there are abnormalities or changes in the natural spinal curvature, these abnormalities are named with the following conditions and include the following symptoms. LORDOSIS Some lordosis is normal in the lower portion or, lumbar section, of the human spine. A decreased or exaggerated amount of lordosis that is causing spinal instability is a condition that may affect some patients. Symptoms of Lordosis include: ● Appearance of sway back where the lower back region has a pronounced curve and looks hollow with a pronounced buttock area ● Difficulty with movement in certain directions ● Low back pain KYPHOSIS This condition is diagnosed when the patient has a rounded upper back and the spine is bent over or curved more than 50 degrees. Symptoms of Kyphosis include: ● Curved or hunched upper back ● Patient’s head that leans forward ● May have upper back pain ● Experiences upper back discomfort after movement or exercise SCOLIOSIS The most common of the three curvatures. This condition is diagnosed when the spine looks like a “s” or “c” from the back. The spine is not straight up and down but has a curve or two running side-to-side. Sagittal Balance Definition • Sagittal= front-to-back direction (sagittal plane) • Imbalance= Lack of harmony or balance Etiology • Excessive lordosis (backwards lean) or kyphosis (forward lean) • Traumatic injury • Previous spinal fusion that disrupted sagittal balance Effects • Low back pain • Difficulty walking • Inability to look straight ahead when upright The most ergonomic and natural posture is to maintain neutral balance, with the head positioned over the shoulders and pelvis. -

Orthotic Treatment of Adult Scoliosis Patients with Chronic Back Pain Dino Gallo
Gallo Scoliosis 2014, 9:18 http://www.scoliosisjournal.com/content/9/1/18 CASE REPORT Open Access Case reports: orthotic treatment of adult scoliosis patients with chronic back pain Dino Gallo Abstract Orthotic treatment of patients with degenerative deformations of the spine is a complex endeavor. It is a great orthopedic technical challenge to effectively reduce the accompanying pain and to help patients regain and keep their mobility. Due to difficult therapies and poor compliance, a surgical intervention to brace the spine is usually the first therapeutic choice. This article presents two cases in which individualized torso orthoses were successfully used to treat patients with degenerative diseases and disorders of the sagittal line as well as three dimensional deformities of the spine. Using torso orthoses allows treatment of these patients with as few invasive measures as possible without losing maximal functionality. Keywords: Scoliosis, Spine deformity, Customized trunk orthoses, Body, Alignment, Chronic back pain, Sagittal line Introduction medically indicated for adult scoliosis patients with chronic Treatment of patients with degenerative deformations of pain if the therapeutic effect of the orthosis is established the spine and of adult scoliosis patients with chronic [3,4]. There are several published reports that show the back pain is challenging. Accompanying orthotic ser- effects of specific physiotherapeutic treatment and the vices, which are mandatory in many cases, are compli- use of elastic [5] and solid braces. Some authors even cated and compliance is often poor. Consequently, a describe a correction of the sagittal profile only by use surgical intervention to brace the spine is the first thera- of an orthosis [1,2]. -

REGROUPI NG Congenital & Pediatric
REGROUPI NG 2 Congenital & Ped iatric CONGENITAL & PAEDIATRIC 18.02.05 Preamble - Objectives and Outcomes ALSO SEE OVERALL PREAMBLE (hypertext link on webpage) Many children and young adults experience congenital health problems which require plastic and/or reconstructive surgery to enable them to function normally. To be effective in this area a surgeon requires technical skill, medical expertise and the capacity to respond effectively to their patients' needs and expectations" The graduating trainee will be able to: • Consistently demonstrate sound surgical skills • Maintain skills and learn new skills • Effectively manage complications • Manage complexity and uncertainty • Appraise and interpret plain radiographs, CT and MRI against patients' needs • Communicate information to patients (and their fa mily) about procedures, potentialities and risks associated with surgery in ways that encourage their participation in informed decision making • Develop a care plan for a patient in collaboration with members of an interdisciplinary team • Promote health maintenance • Draw on different kinds of knowledge in order to weigh up patient's problems in terms of context, issues, needs and consequences For Recommended Reading, Delivery and Assessment see the module fo r each body zone Revisional Knowledge following on from that gained from the PRS Science and Principles Module trainees are required to be able to analyse and appropriately apply the science and principles of the following in clinical environments : Craniomaxillofacial Cra niomaxillofacial embryology, anatomy, genetics • Pathogenesis of craniofacial clefts and their classification • Perioperative management of neurosurgical/orbital surgical/major facial surgical patients (including paediatric) Trunk, Perineum & Breast Embryology • Urogenital embryology - male, female, androgenic influence • Breast embryology Congenital Defects and their cla ssification • Spina bifida • Gastroschisis, omphalocele, Prune-belly • Pectus excavatum, pectus carinatum, Poland syndrome . -

Megalencephaly and Macrocephaly
277 Megalencephaly and Macrocephaly KellenD.Winden,MD,PhD1 Christopher J. Yuskaitis, MD, PhD1 Annapurna Poduri, MD, MPH2 1 Department of Neurology, Boston Children’s Hospital, Boston, Address for correspondence Annapurna Poduri, Epilepsy Genetics Massachusetts Program, Division of Epilepsy and Clinical Electrophysiology, 2 Epilepsy Genetics Program, Division of Epilepsy and Clinical Department of Neurology, Fegan 9, Boston Children’s Hospital, 300 Electrophysiology, Department of Neurology, Boston Children’s Longwood Avenue, Boston, MA 02115 Hospital, Boston, Massachusetts (e-mail: [email protected]). Semin Neurol 2015;35:277–287. Abstract Megalencephaly is a developmental disorder characterized by brain overgrowth secondary to increased size and/or numbers of neurons and glia. These disorders can be divided into metabolic and developmental categories based on their molecular etiologies. Metabolic megalencephalies are mostly caused by genetic defects in cellular metabolism, whereas developmental megalencephalies have recently been shown to be caused by alterations in signaling pathways that regulate neuronal replication, growth, and migration. These disorders often lead to epilepsy, developmental disabilities, and Keywords behavioral problems; specific disorders have associations with overgrowth or abnor- ► megalencephaly malities in other tissues. The molecular underpinnings of many of these disorders are ► hemimegalencephaly now understood, providing insight into how dysregulation of critical pathways leads to ► -

Non-Cardiac Manifestations of Marfan Syndrome
Keynote Lecture Series Non-cardiac manifestations of Marfan syndrome Anne H. Child Molecular and Clinical Sciences Research Institute, St George’s University of London, Cranmer Terrace, London, UK Correspondence to: Dr. Anne H. Child, MD, FRCP. Reader in Cardiovascular Genetics, Molecular and Clinical Sciences Research Institute, St George's University of London, Cranmer Terrace, London SW17 0RE, UK. Email: [email protected]. Because of the widespread distribution of fibrillin 1 in the body, Marfan syndrome (MFS) affects virtually every system. The expression of this single dominantly inherited gene is variable within a family, and between families. There is some genotype-phenotype correlation which is helpful in guiding long-term prognosis, and management. In general gene mutations have been reported in clusters, with those having mainly ocular manifestations occurring in exons 1 to 15 of this 65-exon gene; those causing cardiac problems often involving cysteine replacement in a calcium binding EGF-like sequence; the most severe mutations occurring in exons 25–32, causing neonatal MFS diagnosed at birth, and severe enough to cause death frequently before the age of 2. Other correlations will certainly be found in future. This condition is progressive, and the manifestations unfold according to age. For example, if the lens is going to dislocate this usually occurs by age 10; scoliosis usually presents itself between the ages of 8 and 15; height should be monitored carefully between the onset of puberty and cessation of growth approximately age 17 or 18. Holistic care should be offered by one doctor who oversees the patient’s welfare. This should be a paediatrician, paediatric cardiologist, or general practitioner in the case of an affected child. -

SCOLIOSIS: What Is New and True? Natural History, Screening/Evaluation and Treatment
SCOLIOSIS: what is new and true? Natural history, screening/evaluation and treatment Kathleen Moen MD Swedish Pediatric Specialty Update January 24, 2020 OBJECTIVES • 1) review of entity adolescent idiopathic scoliosis • 2) review the natural history of treated and untreated disease • 3) review screening recommendations and delineating patients who warrant referral • 4) review of treatment modalities Scoliosis • Definition: a lateral spinal curvature measuring at least 10 degrees on xray • Complex 3dimensional spinal deformity Adolescent Idiopathic Scoliosis • AIS: Scoliosis most often develops and progresses during the most rapid times of growth, typically between ages 10 ‐15 years. • NOT infantile or juvenile scoliosis which have different natural history profiles • NOT congenital spinal deformities, neuromuscular scoliosis or spinal dysraphism Adolescent Idiopathic Scoliosis • Classic Teaching – Small curves are common – Progression occurs during most rapid times of growth – Female predominance – Big curves get bigger – Etiology unclear: familial predisposition, and? – Idiopathic scoliosis not typically painful Adolescent Idiopathic Scoliosis • Mild scoliosis curves are common, affecting 2‐3% of the adolescent population, males and females equally • Less than 0.1% of adolescents have curves greater than 40 degrees. • Ratio of females to males 10:1 Cobb Angle Female:Male Prevalence >10 1.4‐2 :1 2 ‐ 3 % >20 5:1 .3 ‐ .5 % >30 10:1 .1 ‐ .3 % >40 10:1 <0.1% Weinstein, SL Adolescent Idiopathic Scoliosis, Prevalence and Natural History. Instructional -

[email protected] Patient Information Patient Name (Last, First, Middle) Birth Date (Mm-Dd-Yyyy) Sex Male Female
Marfan and Related Disorders Patient Information Instructions: The accurate interpretation and reporting of the genetic results is contingent upon the reason for referral, clinical information, ethnic background, and family history. To help provide the best possible service, supply the information requested below and send paperwork with the specimen, or return by fax to Mayo Clinic Laboratories, Attn: Personalized Genomics Laboratory Genetic Counselors at 507-284-1759. Phone: 507-266-5700 / International clients: +1-507-266-5700 or email [email protected] Patient Information Patient Name (Last, First, Middle) Birth Date (mm-dd-yyyy) Sex Male Female Referring Provider Name (Last, First) Phone Fax* Other Contact Name (Last, First) Phone Fax* *Fax number given must be from a fax machine that complies with applicable HIPAA regulations. Is this a postmortem specimen? Yes No If yes, attach autopsy report if available. Clinical History Reason for Testing (Check all that apply.) Diagnosis Carrier testing Presymptomatic diagnosis Family history Sudden death Note: Genetic testing should always be initiated on an affected family member first, if possible, in order to be most informative for at-risk relatives. See Ethnic Background and Family History section for more information. Diagnosis/Suspected Diagnosis Marfan Syndrome Ehlers-Danlos Syndrome Loeys-Dietz Syndrome Familial thoracic aortic aneurysm and dissection Other: _______________________________________________________________________________________________ Indicate whether the following -

Kyphectomy in Neonates with Meningomyelocele
Child's Nervous System (2019) 35:673–681 https://doi.org/10.1007/s00381-018-4006-4 ORIGINAL PAPER Kyphectomy in neonates with meningomyelocele Nail Özdemir1 & Senem Alkan Özdemir2 & Esra Arun Özer3 Received: 18 August 2018 /Accepted: 18 November 2018 /Published online: 11 December 2018 # Springer-Verlag GmbH Germany, part of Springer Nature 2018 Abstract Purpose Kyphosis is the most severe spinal deformity associated with meningomyelocele (MMC) and is seen in approximately 15% of neonates. Our purpose is to present our clinical experience, to discuss the technique and deformity correction in kyphectomy in neonates with MMC, and to assess its long-term outcomes. Method In this prospective study, the authors reviewed eight cases submitted to surgery between 2013 and 2015. We evaluated clinical characteristics that were analyzed, as were the operative technique employed, and angle range of the kyphosis deformity postcorrection follow-up. Results Neonatal kyphectomy was performed of six females and two males. The mean birth weight was 2780 g, and the mean age at the time of surgery was 5.6 days. There were S-shaped type deformity in lumbar region in all neonates. In the correction of the kyphotic deformity, a total vertebrae were removed from four patient, whereas a partial vertebrectomy was done in four. The mean operative time was 116 min. No patients did not require the blood transfusion. There were no serious complications, and wound closure was successful in all patients. The mean follow-up period was 4 years and 3 months (range 36–61 months), except one patient who died 1 week after discharge.