Developmental Abnormalities Associated with a Ring
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genetical Society of Great Britain
Heredity 59 (1987) 151—160 The Genetical Society of Great Britain THEGENETICAL SOCIETY (Abstracts of papers presented at the TVVO HUNDRED AND FIFTH MEETING of the Society held on Friday, 14th and Saturday, 15th November 1986 at UNIVERSITY COLLEGE, LONDON) 1. Selection of somatic cell D. J. Porteous, P. A. Boyd, N. D. Hastie and hybrids with specific chromosome V. van Heyningen content for mapping the WAGR MAC Clinical and Population Cytogenetics Unit, Western General Hospital, Crewe Road, syndrome Edinburgh EH4 2XU. J. M. Fletcher, H. Morrison, J. A. Fantes, Clonedprobes for a number of available chromo- A. Seawright, S. Christie, D. J. Porteous, some ii assigned genes were used to define the N. D. Hastie and V. van Heyningen extent of deletions associated with the Wilms' MAC Clinical and Population Cytogenetics Unit, tumour, aniridia, genitourinary abnormalities and Western General Hospital, Crewe Road, mental retardation (WAGR) syndrome. Establish- Edinburgh EH4 2XU. ing reliable dosage studies for a number of different probes has proved difficult. We have therefore WAGR(Wilms tumour, aniridia, genitourinary abnormalities and mental retardation) syndrome concentrated on segregating the deleted chromo- is frequently associated with deletions on the short some 11 from a number of patients in somatic cell arm of chromosome 11. The deletions vary in size hybrids and analysing DNA from these to produce but always include part of band lipl3. To home a consistent map of chromosome lip. At the same in on the Wilms tumour and aniridia loci the end time we have determined the deletion breakpoints points of the different deletion breakpoints need at a molecular level and shown that the results are to be defined at the DNA level. -

Ring Chromosome 4 49,XXXXY Patients Is Related to the Age of the Mother
228 Case reports placenta and chorionic sacs were of no help for Further cytogenetic studies in twins would be diagnosis. The dermatoglyphs are expected to be necessary to find out whether there is a relation different, even ifthey were monozygotic, in relation to between non-disjunction and double ovulation or the total finger ridge count; since according to whether these 2 events are independent but could Penrose (1967), when the number of X chromosomes occur at the same time by chance. increases, the TFRC decreases in about 30 per each extra X. The difference of 112 found in our case is so We want to thank Dr Maroto and Dr Rodriguez- striking that we believe that we are facing a case of Durantez for performing the cardiological and dizygosity. On the other hand, the blood groups were radiological studies; Dr A. Valls for performing the conclusive. All the systems studied were alike in Xg blood group. We also wish to thank Mrs A. both twins except for the Rh. In the propositus the Moran and Mrs M. C. Cacituaga for their technical phenotype was CCDee while in the brother it was assistance. cCDee, which rules out monozygosity. The incidence of dizygotic twins with noncon- J. M. GARCIA-SAGREDO, C. MERELLO-GODINO, cordant chromosomal aneuploidy appears to be low. and C. SAN ROMAN To the best of our knowledge we think that ours is the From the Department ofHuman Genetics, first reported case of dizygotic twins with this specific Fundacion Jimenez Diaz, Madrid; and anomaly. U.C.I., Hospital Infantil, C.S. -

Ring 21 FTNW
Ring 21 rarechromo.org Sources Ring 21 The information Ring 21 is a rare genetic condition caused by having a in this leaflet ring-shaped chromosome. comes from the Almost half of the people with ring 21 chromosomes medical literature described in the medical literature are healthy and and from develop normally. Their unusual chromosomes are Unique’s discovered by chance, during tests for infertility or after members with repeated miscarriages or after having an affected baby. Ring 21 In other people the ring 21 chromosome affects (referenced U), development and learning and can also cause medical who were problems. In most of these people these effects are surveyed in slight but in some people they can be severe. The 2004. Unique is effects can even vary between different members of the very grateful to same family. The reason for these differences is not yet the families who fully understood. took part in the survey. What is a chromosome? The human body is made up of cells. Inside most cells is References a nucleus where genetic information is stored in genes which are grouped along chromosomes. Chromosomes The text contains are large enough to be studied under a microscope and references to come in different sizes, each with a short (p) and a long articles published (q) arm. They are numbered from largest to smallest in the medical according to their size, from number 1 to number 22, in press. The first- addition to the sex chromosomes, X and Y. A normal, named author healthy cell in the body has 46 chromosomes, 23 from and publication the mother and 23 from the father, including one date are given to chromosome 21 from each parent. -

22Q13.3 Deletion Syndrome
22q13.3 deletion syndrome Description 22q13.3 deletion syndrome, which is also known as Phelan-McDermid syndrome, is a disorder caused by the loss of a small piece of chromosome 22. The deletion occurs near the end of the chromosome at a location designated q13.3. The features of 22q13.3 deletion syndrome vary widely and involve many parts of the body. Characteristic signs and symptoms include developmental delay, moderate to profound intellectual disability, decreased muscle tone (hypotonia), and absent or delayed speech. Some people with this condition have autism or autistic-like behavior that affects communication and social interaction, such as poor eye contact, sensitivity to touch, and aggressive behaviors. They may also chew on non-food items such as clothing. Less frequently, people with this condition have seizures or lose skills they had already acquired (developmental regression). Individuals with 22q13.3 deletion syndrome tend to have a decreased sensitivity to pain. Many also have a reduced ability to sweat, which can lead to a greater risk of overheating and dehydration. Some people with this condition have episodes of frequent vomiting and nausea (cyclic vomiting) and backflow of stomach acids into the esophagus (gastroesophageal reflux). People with 22q13.3 deletion syndrome typically have distinctive facial features, including a long, narrow head; prominent ears; a pointed chin; droopy eyelids (ptosis); and deep-set eyes. Other physical features seen with this condition include large and fleshy hands and/or feet, a fusion of the second and third toes (syndactyly), and small or abnormal toenails. Some affected individuals have rapid (accelerated) growth. -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Chromosome 2 Introduction the Genetic Length of Chromosome 2
Chromosome 2 ©Chromosome Disorder Outreach Inc. (CDO) Technical genetic content provided by Dr. Iosif Lurie, M.D. Ph.D Medical Geneticist and CDO Medical Consultant/Advisor. Ideogram courtesy of the University of Washington Department of Pathology: ©1994 David Adler.hum_02.gif Introduction The genetic length of chromosome 2 spans ~237 Mb. It is ~8% of the whole human genetic material. The length of the short arm is ~89 Mb; the length of the long arm is 148 Mb. Chromosome 2 contains ~1500 genes. At least 200 of these genes are related to different kinds of genetic pathologies. Many of these genes are associated with structural defects of the organs, and others, to functional defects. The numerous structural abnormalities of chromosome 2 had already been reported before methods of molecular genetics gained wide usage. Currently there are ~1,500 reports on patients with varying structural abnormalities of this chromosome, including ~1,000 reports on patients with deletions. Deletions have been reported in at least 10–15 patients for each segment of chromosome 2. There are ten known syndromes caused by deletions of chromosome 2, including three syndromes related to deletions of the short arm (p). Some of these syndromes have been known for many years; others (del 2p15p16.1, del 2q23q24, and del 2q32) have been delineated only in the last couple of years. Deletions of Chromosome 2 Different types of chromosome 2 deletions have been described in numerous publications and may be subdivided into 3 groups: • Deletions that are “harmless” variants (usually familial) • Unique deletions that have not been categorized as a syndrome yet • Deletions that are considered a syndrome Deletions of 2p Deletion of 2p15p16.1 This syndrome, first described in 2007, is rare; only ten patienys have been described to date. -

Chromosome 6 Abnormalities in Ovarian Surface Epithelial Tumors of Borderline Malignancy Suggest a Genetic Continuum in the Progression Model of Ovarian Neoplasms1
3404 Vol. 7, 3404–3409, November 2001 Clinical Cancer Research Chromosome 6 Abnormalities in Ovarian Surface Epithelial Tumors of Borderline Malignancy Suggest a Genetic Continuum in the Progression Model of Ovarian Neoplasms1 Maria Grazia Tibiletti, Barbara Bernasconi, Conclusion: Alterations in the above-mentioned inter- Daniela Furlan, Paola Bressan, Roberta Cerutti, val are a common finding in advanced ovarian carcinomas but also in benign ovarian cysts, implying that some tumors Carla Facco, Massimo Franchi, Cristina Riva, of borderline malignancy may arise from benign tumors and Raffaella Cinquetti, Carlo Capella, and that malignant ones may evolve from tumors of borderline Roberto Taramelli2 malignancy. Genes located in 6q27 seem to be crucial for this Laboratorio di Anatomia Patologica Ospedale di Circolo, Fondazione mechanism of early events in ovarian tumorigenesis. Macchi, 21100 Varese [M. G. T., C. F., C. R.]; and Dipartimento di Scienze Cliniche e Biologiche [B. B., D. F., P. B., R. Ce., C. C.], INTRODUCTION Clinica Ostetrica e Ginecologica [M. F.], and Dipartimento di Biologia Strutturale e Funzionale [R. Ci., R. T.], Universita` Ovarian cancer represents the most lethal of the gyneco- dell’Insubria, 21100 Varese, Italy logical tumors, and its pathogenesis is poorly understood. More than 80% of malignant tumors of the ovary are of epithelial origin and arise from the surface epithelium (1). Ovarian surface ABSTRACT epithelial tumors are divided into three histological subtypes: Purpose: We used conventional cytogenetics, molecular benign cystoadenoma, TBM,3 and AC, which are related to a cytogenetics, and molecular genetic analyses to study the different biological behavior (1). The molecular events under- pattern of allelic loss on chromosome 6q in a cohort of lying the development of ovarian tumorigenesis are still largely borderline epithelial ovarian tumors. -
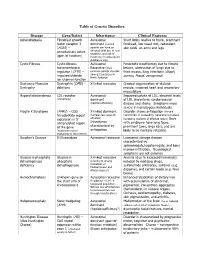
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical
Table of Genetic Disorders Disease Gene/Defect Inheritance Clinical Features Achondroplasia Fibroblast growth Autosomal Short limbs relative to trunk, prominent factor receptor 3 dominant (normal forehead, low nasal root, redundant (FGR3) – parents can have an skin folds on arms and legs constitutively active affected child due to new mutation, and risk of (gain of function) recurrence in subsequent children is low) Cystic Fibrosis Cystic fibrosis Autosomal Pancreatic insufficiency due to fibrotic transmembrane Recessive (most lesions, obstruction of lungs due to regulator (CFTR) – common genetic disorder thick mucus, lung infections (Staph, impaired chloride among Caucasians in aureus, Pseud. aeruginosa) North America) ion channel function Duchenne Muscular Dystrophin (DMD) - X-linked recessive Gradual degeneration of skeletal Dystrophy deletions muscle, impaired heart and respiratory musculature Hypercholesterolemia LDL receptor Autosomal Impaired uptake of LDL, elevated levels (commonly) dominant of LDL cholesterol, cardiovascular (haploinsufficiency) disease and stroke. Symptoms more severe in homozygous individuals Fragile X Syndrome (FMR1) – CGG X-linked dominant Disorder shows anticipation (female trinucleotide repeat (females less severely transmitters in succeeding generations produce expansion in 5’ affected) increasing numbers of affected males) Boys untranslated region Inheritance with syndrome have long faces, of the gene characterized by prominent jaws, large ears, and are (expansion occurs anticipation likely to be mentally retarded. exclusively in the mother) Gaucher’s Disease Β-Glucosidase Autosomal recessive Lysosomal storage disease characterized by splenomegaly,hepatomegaly, and bone marrow infiltration. Neurological symptoms are not common Glucose 6-phosphate Glucose 6- X-linked recessive Anemia (due to increased hemolysis) dehydrogenase phosphate (prominent among induced by oxidizing drugs, deficiency dehydrogenase individuals of sulfonamide antibiotics, sulfones (e.g. -

Inside This Issue
Winter 2014 No. 77 Inside this issue Group News | Fundraising | Members’ Letters | One Family Living with Two Different Chromosome Disorders | Bristol Conference 2014 | Unique Leaflets | Christmas Card Order Form Sophie, Unique’s Chair of Trustees Dear Members, In the past month a few things have reminded me of why it is so important to make connections through Unique but also to draw support from other parents around us. I’ve just returned from Unique’s most recent family conference in Bristol where 150 of us parents and carers had a lovely time in workshops, meals and activities, chatting and watching our children milling around together like one big family since – although we had never met before – we have shared so many experiences in common. However in contrast I have also just met a new mum who has just moved to my area from far away with two toddlers, one with a rare joys of the internet, it is becoming easier to meet others with similar, chromosome disorder, who is starting from scratch with no even very rare, chromosome disorders around the world and to find professional, medical or social support. She reminds me of how yourself talking to them in the middle of the night about some lonely I felt when Max was newly diagnosed, when I knew no one interesting things our children share in common (obsession with with a disabled child let alone anyone with a rare chromosome catalogues, anyone?) And of course we also have an enormous disorder. Elsewhere our latest Unique Facebook group, Unique amount in common with so many parents of children with other Russia, is also just starting up – so far it includes just a small special needs or disabilities around us in our own communities who number of members sharing very different experiences to mine here will often be walking the same path as us. -

Molecular Cytogenetic Characterisation of a Novel De Novo Ring
Pace et al. Molecular Cytogenetics (2017) 10:9 DOI 10.1186/s13039-017-0311-y CASEREPORT Open Access Molecular cytogenetic characterisation of a novel de novo ring chromosome 6 involving a terminal 6p deletion and terminal 6q duplication in the different arms of the same chromosome Nikolai Paul Pace1, Frideriki Maggouta2, Melissa Twigden2 and Isabella Borg1,3,4* Abstract Background: Ring chromosome 6 is a rare sporadic chromosomal abnormality, associated with extreme variability in clinical phenotypes. Most ring chromosomes are known to have deletions on one or both chromosomal arms. Here, we report an atypical and unique ring chromosome 6 involving both a distal deletion and a distal duplication on the different arms of the same chromosome. Case presentation: In a patient with intellectual disability, short stature, microcephaly, facial dysmorphology, congenital heart defects and renovascular disease, a ring chromosome 6 was characterised using array-CGH and dual-colour FISH. The de-novo ring chromosome 6 involved a 1.8 Mb terminal deletion in the distal short arm and a 2.5 Mb duplication in the distal long arm of the same chromosome 6. This results in monosomy for the region 6pter to 6p25.3 and trisomy for the region 6q27 to 6qter. Analysis of genes in these chromosomal regions suggests that haploinsufficiency for FOXC1 and GMDS genes accounts for the cardiac and neurodevelopmental phenotypes in the proband. The ring chromosome 6 reported here is atypical as it involves a unique duplication of the distal long arm. Furthermore, the presence of renovascular disease is also a unique feature identified in this patient. -

Poster Presentations in Cytogenetics
Poster Presentations in Cytogenetics Trisomy 8 in cervical cancer. D. Feldman. S. Das. H. Kve. C:L. Sun. !vL Mosaicism for duplication of 17q21 .qter with lymphedema and normal phenotype. M. Descartes. L. Baldwln. P. Cosper. A. Carroll. Department Samv and H. F. L. Mark. Lifespan Academic Medical Center Cytogenetics Laboratory, Rhode Island Hospital and Brown University School of of Human Genetics, University of Alabama at Birmingham, Alabama. Medicine, Providence, R1. Duplication of 17q21 .qter is associated with a clinically recognizable Cervical cancer is a malignancy which typically occurs at the syndrome. The major features are, profound mental retardation; dwartism; transformation zone between squamous and glandular epithelium. The vast frontal bossing and temporal retraction, narrowing of the eyes; thln lips wlth malorlty fall into two histologic types: squamous cell and adenocarcinoma. overlapping of the lower lip by the upper lip; abnormal ears; cleft palate' We have previously reported extensively on abnormal chromosome 8 copy The region that appears to be respons~blefor the phenotype Is number in varlous cancers, wh~chappears to be an ubiquitous phenomenon. 17q23 .qterl Serothken et al, reported an infant mosaic for the duplication In the present pilot project, we studied chromosome 8 copy number together 17q21.1 -qter, their patient had many features suggestive of the 17q with a chromosome 17 control using formalin-fixed paraffin-embedded duplications syndrome except for the craniofacial dysmorphism3. We arch~valcervlcal cancer tissues. HER-2/neu oncogene amplification was report an infant who was found to be mosaic for duplication 17q21 .qter also studied in this sample, as reported in a previous abstract presented at who had none of the features associated wlth thls syndrome the 1998 Annual Meeting of the Amencan Society of Human Genetics. -

The Cytogenetics of Hematologic Neoplasms 1 5
The Cytogenetics of Hematologic Neoplasms 1 5 Aurelia Meloni-Ehrig that errors during cell division were the basis for neoplastic Introduction growth was most likely the determining factor that inspired early researchers to take a better look at the genetics of the The knowledge that cancer is a malignant form of uncon- cell itself. Thus, the need to have cell preparations good trolled growth has existed for over a century. Several biologi- enough to be able to understand the mechanism of cell cal, chemical, and physical agents have been implicated in division became of critical importance. cancer causation. However, the mechanisms responsible for About 50 years after Boveri’s chromosome theory, the this uninhibited proliferation, following the initial insult(s), fi rst manuscripts on the chromosome makeup in normal are still object of intense investigation. human cells and in genetic disorders started to appear, fol- The fi rst documented studies of cancer were performed lowed by those describing chromosome changes in neoplas- over a century ago on domestic animals. At that time, the tic cells. A milestone of this investigation occurred in 1960 lack of both theoretical and technological knowledge with the publication of the fi rst article by Nowell and impaired the formulations of conclusions about cancer, other Hungerford on the association of chronic myelogenous leu- than the visible presence of new growth, thus the term neo- kemia with a small size chromosome, known today as the plasm (from the Greek neo = new and plasma = growth). In Philadelphia (Ph) chromosome, to honor the city where it the early 1900s, the fundamental role of chromosomes in was discovered (see also Chap.