Decoding the Fine-Scale Structure of a Breast Cancer Genome and Transcriptome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
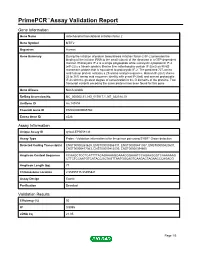
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name mitochondrial translational initiation factor 2 Gene Symbol MTIF2 Organism Human Gene Summary During the initiation of protein biosynthesis initiation factor-2 (IF-2) promotes the binding of the initiator tRNA to the small subunit of the ribosome in a GTP-dependent manner. Prokaryotic IF-2 is a single polypeptide while eukaryotic cytoplasmic IF-2 (eIF-2) is a trimeric protein. Bovine liver mitochondria contain IF-2(mt) an 85-kD monomeric protein that is equivalent to prokaryotic IF-2. The predicted 727-amino acid human protein contains a 29-amino acid presequence. Human IF-2(mt) shares 32 to 38% amino acid sequence identity with yeast IF-2(mt) and several prokaryotic IF-2s with the greatest degree of conservation in the G domains of the proteins. Two transcript variants encoding the same protein have been found for this gene. Gene Aliases Not Available RefSeq Accession No. NC_000002.11, NG_017017.1, NT_022184.15 UniGene ID Hs.149894 Ensembl Gene ID ENSG00000085760 Entrez Gene ID 4528 Assay Information Unique Assay ID qHsaCEP0058136 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000263629, ENST00000366137, ENST00000441307, ENST00000420637, ENST00000417363, ENST00000412530, ENST00000394600 Amplicon Context Sequence CCAAGCTCCTCATTTTACAGAAAAGGAAACGGAAATCCAGAAGGGTCAAAAAAG CTTCTCCAATGTCATACCGCTAGTTAATGGCAGTCAAGACTAGAACCCAGACG Amplicon Length (bp) 77 Chromosome Location 2:55495715-55495821 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 95 R2 0.9995 cDNA Cq 21.05 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79 gDNA Cq 24.56 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

C. Elegans Whole Genome Sequencing Reveals Mutational Signatures Related to Carcinogens and DNA Repair Deficiency
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press C. elegans whole genome sequencing reveals mutational signatures related to carcinogens and DNA repair deficiency Authors: Bettina Meier * (1); Susanna L Cooke * (2); Joerg Weiss (1); Aymeric P Bailly (1,3); Ludmil B Alexandrov (2); John Marshall (2); Keiran Raine (2); Mark Maddison (2); Elizabeth Anderson (2); Michael R Stratton (2); Anton Gartner * (1); Peter J Campbell * (2,4,5). * These authors contributed equally to this project. Institutions: (1) Centre for Gene Regulation and Expression, University of Dundee, Dundee, UK. (2) Cancer Genome Project, Wellcome Trust Sanger Institute, Hinxton, UK. (3) CRBM/CNRS UMR5237, University of Montpellier, Montpellier, France. (4) Department of Haematology, University of Cambridge, Cambridge, UK. (5) Department of Haematology, Addenbrooke’s Hospital, Cambridge, UK. Address for correspondence: Dr Peter J Campbell, Dr Anton Gartner, Cancer Genome Project, Centre for Gene Regulation and Expression, Wellcome Trust Sanger Institute, The University of Dundee, Hinxton CB10 1SA, Dow Street, Cambridgeshire, Dundee DD1 5EH UK. UK. Tel: +44 (0) 1223 494745 Phone: +44 (0) 1382 385809 Fax: +44 (0) 1223 494809 E-mail: [email protected] E-mail: [email protected] Running title: mutation profiling in C. elegans Keywords: mutation pattern, genetic and environmental factors, C. elegans, cisplatin, aflatoxin B1, whole-genome sequencing. Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Mutation is associated with developmental and hereditary disorders, ageing and cancer. While we understand some mutational processes operative in human disease, most remain mysterious. -

Abstracts In
ECCB 2014 Accepted Posters with Abstracts G: Bioinformatics of health and disease G01: Emile Rugamika Chimusa, Jacquiline Wangui Mugo and Nicola Mulder. Leveraging ancestry along the genome of admixed individuals to resolve missing heritability in disease scoring statistics Abstract: Human genetics has been haunted by the mystery of “missing heritability” of common traits. Although studies have discovered several variants associated with common diseases and traits, these variants typically appear to explain only a minority of the heritability. Resolving missing heritability, the difference between phenotypic variance explained by associated SNPs and estimates of narrow-sense heritability (h2), will inform strategies for disease mapping and prediction of complex traits. Among biased estimates of h2 due to epistatic interactions and rare variants not captured by genotyping arrays have been cited to be the most can be the most explanations for missing heritability. Here, we present an approach for estimating heritability of traits based on sharing local ancestry segments between pairs of unrelated individuals in an admixed population. From simulation data and real data, we demonstrated that our approach outperformed current approaches for estimating heritability of traits and holds values in admixture mapping for deconvoluting genes underlying ethnic differences in complex diseases risk. G02: Sylvain Mareschal, Pierre-Julien Viailly, Philippe Bertrand, Fabienne Desmots-Loyer, Elodie Bohers, Catherine Maingonnat, Karen Leroy, Thierry Fest and Fabrice Jardin. Next- Generation Sequencing applied to tailor targeted therapies in lymphoma: the RELYSE project Abstract: Non-Hodgkin Lymphomas (NHL) are lymphoid cell malignancies accounting for about 4% of all cancers, with an incidence rate of 12 cases per 100,000 and per year in Europe. -

Role of Phytochemicals in Colon Cancer Prevention: a Nutrigenomics Approach
Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Marjan J van Erk Promotor: Prof. Dr. P.J. van Bladeren Hoogleraar in de Toxicokinetiek en Biotransformatie Wageningen Universiteit Co-promotoren: Dr. Ir. J.M.M.J.G. Aarts Universitair Docent, Sectie Toxicologie Wageningen Universiteit Dr. Ir. B. van Ommen Senior Research Fellow Nutritional Systems Biology TNO Voeding, Zeist Promotiecommissie: Prof. Dr. P. Dolara University of Florence, Italy Prof. Dr. J.A.M. Leunissen Wageningen Universiteit Prof. Dr. J.C. Mathers University of Newcastle, United Kingdom Prof. Dr. M. Müller Wageningen Universiteit Dit onderzoek is uitgevoerd binnen de onderzoekschool VLAG Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Marjan Jolanda van Erk Proefschrift ter verkrijging van graad van doctor op gezag van de rector magnificus van Wageningen Universiteit, Prof.Dr.Ir. L. Speelman, in het openbaar te verdedigen op vrijdag 1 oktober 2004 des namiddags te vier uur in de Aula Title Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Author Marjan Jolanda van Erk Thesis Wageningen University, Wageningen, the Netherlands (2004) with abstract, with references, with summary in Dutch ISBN 90-8504-085-X ABSTRACT Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Specific food compounds, especially from fruits and vegetables, may protect against development of colon cancer. In this thesis effects and mechanisms of various phytochemicals in relation to colon cancer prevention were studied through application of large-scale gene expression profiling. Expression measurement of thousands of genes can yield a more complete and in-depth insight into the mode of action of the compounds. -

Human Genetics: International Projects and Personalized Medicine
Drug Metabol Pers Ther 2016; 31(1): 3–8 Mini Review Maria Apellaniz-Ruiza, Cristina Gallegoa, Sara Ruiz-Pintoa, Angel Carracedo and Cristina Rodríguez-Antona* Human genetics: international projects and personalized medicine DOI 10.1515/dmpt-2015-0032 Received August 31, 2015; accepted October 19, 2015; previously Introduction published online November 18, 2015 Genetic variation databases describe naturally occur- Abstract: In this article, we present the progress driven ring genetic differences among individuals of the same by the recent technological advances and new revolu- species. This variation, accounting for 0.1% of our DNA tionary massive sequencing technologies in the field of [1], permits the flexibility and survival of a population human genetics. We discuss this knowledge in relation in the face of changing environmental circumstances, with drug response prediction, from the germline genetic but it also influences how people differ in their risk of variation compiled in the 1000 Genomes Project or in the disease or their response to drugs. It is well known that Genotype-Tissue Expression project, to the phenome- variability in response to drug therapy is the rule rather genome archives, the international cancer projects, such than the exception for most drugs, and these differences as The Cancer Genome Atlas or the International Cancer are among the major challenges in current clinical prac- Genome Consortium, and the epigenetic variation and its tice, drug development, and drug regulation [2, 3]. Thus, influence in gene expression, including the regulation of rather than accepting the “one drug fits all” approach, drug metabolism. This review is based on the lectures pre- researchers envision that drugs need to be tailored to fit sented by the speakers of the Symposium “Human Genet- the profile of each individual patient. -

By Submitted in Partial Satisfaction of the Requirements for Degree of in In
BCL6 maintains thermogenic capacity of brown adipose tissue during dormancy by Vassily Kutyavin DISSERTATION Submitted in partial satisfaction of the requirements for degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________Eric Verdin Chair ______________________________________________________________________________Ajay Chawla ______________________________________________________________________________Ethan Weiss ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2019 by Vassily Kutyavin ii Dedicated to everyone who has supported me during my scientific education iii Acknowledgements I'm very grateful to my thesis adviser, Ajay Chawla, for his mentorship and support during my dissertation work over the past five years. Throughout my time in his lab, I was always able to rely on his guidance, and his enthusiasm for science was a great source of motivation. Even when he was traveling, he could easily be reached for advice by phone or e- mail. I am particularly grateful for his help with writing the manuscript, which was probably the most challenging aspect of graduate school for me. I am also very grateful to him for helping me find a postdoctoral fellowship position. Ajay's inquisitive and fearless approach to science have been a great inspiration to me. In contrast to the majority of scientists who focus narrowly on a specific topic, Ajay pursued fundamental questions across a broad range of topics and was able to make tremendous contributions. My experience in his lab instilled in me a deep appreciation for thinking about the entire organism from an evolutionary perspective and focusing on the key questions that escape the attention of the larger scientific community. As I move forward in my scientific career, there is no doubt that I will rely on him as a role model. -

Strategic Plan 2011-2016
Strategic Plan 2011-2016 Wellcome Trust Sanger Institute Strategic Plan 2011-2016 Mission The Wellcome Trust Sanger Institute uses genome sequences to advance understanding of the biology of humans and pathogens in order to improve human health. -i- Wellcome Trust Sanger Institute Strategic Plan 2011-2016 - ii - Wellcome Trust Sanger Institute Strategic Plan 2011-2016 CONTENTS Foreword ....................................................................................................................................1 Overview .....................................................................................................................................2 1. History and philosophy ............................................................................................................ 5 2. Organisation of the science ..................................................................................................... 5 3. Developments in the scientific portfolio ................................................................................... 7 4. Summary of the Scientific Programmes 2011 – 2016 .............................................................. 8 4.1 Cancer Genetics and Genomics ................................................................................ 8 4.2 Human Genetics ...................................................................................................... 10 4.3 Pathogen Variation .................................................................................................. 13 4.4 Malaria -

Stroke Genetics and Genomics
Faculdade de Medicina da Universidade de Lisboa Unidade Neurológica de Investigação Clínica PhD Thesis Stroke Genetics and Genomics Tiago Krug Coelho Host Institution: Instituto Gulbenkian de Ciência Supervisor at Instituto Gulbenkian de Ciência: Doctor Sofia Oliveira Supervisor at Faculdade de Medicina da Universidade de Lisboa: Professor José Ferro PhD in Biomedical Sciences Specialization in Neurosciences 2010 Stroke Genetics and Genomics A ciência tem, de facto, um único objectivo: a verdade. Não esgota perfeitamente a sua tarefa se não descobre a causa do todo. Chiara Lubich i Stroke Genetics and Genomics ii Stroke Genetics and Genomics A impressão desta dissertação foi aprovada pela Comissão Coordenadora do Conselho Científico da Faculdade de Medicina de Lisboa em reunião de 28 de Setembro de 2010. iii Stroke Genetics and Genomics iv Stroke Genetics and Genomics As opiniões expressas são da exclusiva responsabilidade do seu autor. v Stroke Genetics and Genomics vi Stroke Genetics and Genomics Abstract ABSTRACT This project presents a comprehensive approach to the identification of new genes that influence the risk for developing stroke. Stroke is the leading cause of death in Portugal and the third leading cause of death in the developed world. It is even more disabling than lethal, and the persistent neurological impairment and physical disability caused by stroke have a very high socioeconomic cost. Moreover, the number of affected individuals is expected to increase with the current aging of the population. Stroke is a “brain attack” cutting off vital blood and oxygen to the brain cells and it is a complex disease resulting from environmental and genetic factors. -

Mitochondrial Genetics
Mitochondrial genetics Patrick Francis Chinnery and Gavin Hudson* Institute of Genetic Medicine, International Centre for Life, Newcastle University, Central Parkway, Newcastle upon Tyne NE1 3BZ, UK Introduction: In the last 10 years the field of mitochondrial genetics has widened, shifting the focus from rare sporadic, metabolic disease to the effects of mitochondrial DNA (mtDNA) variation in a growing spectrum of human disease. The aim of this review is to guide the reader through some key concepts regarding mitochondria before introducing both classic and emerging mitochondrial disorders. Sources of data: In this article, a review of the current mitochondrial genetics literature was conducted using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/). In addition, this review makes use of a growing number of publically available databases including MITOMAP, a human mitochondrial genome database (www.mitomap.org), the Human DNA polymerase Gamma Mutation Database (http://tools.niehs.nih.gov/polg/) and PhyloTree.org (www.phylotree.org), a repository of global mtDNA variation. Areas of agreement: The disruption in cellular energy, resulting from defects in mtDNA or defects in the nuclear-encoded genes responsible for mitochondrial maintenance, manifests in a growing number of human diseases. Areas of controversy: The exact mechanisms which govern the inheritance of mtDNA are hotly debated. Growing points: Although still in the early stages, the development of in vitro genetic manipulation could see an end to the inheritance of the most severe mtDNA disease. Keywords: mitochondria/genetics/mitochondrial DNA/mitochondrial disease/ mtDNA Accepted: April 16, 2013 Mitochondria *Correspondence address. The mitochondrion is a highly specialized organelle, present in almost all Institute of Genetic Medicine, International eukaryotic cells and principally charged with the production of cellular Centre for Life, Newcastle energy through oxidative phosphorylation (OXPHOS). -

Germline Determinants of the Somatic Mutation Landscape in 2,642 Cancer Genomes
bioRxiv preprint doi: https://doi.org/10.1101/208330; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Germline determinants of the somatic mutation landscape in 2,642 cancer genomes Sebastian M Waszak1*, Grace Tiao2*, Bin Zhu3*, Tobias Rausch1*, Francesc Muyas4,5#, Bernardo Rodríguez-Martín6,7#, Raquel Rabionet4#, Sergei Yakneen1#, Georgia Escaramis4,8, Yilong Li9, Natalie Saini10, Steven A Roberts11, German M Demidov4,5, Esa Pitkänen1; Olivier Delaneau12-14, Jose Maria Heredia-Genestar15, Joachim Weischenfeldt1,16, Suyash S Shringarpure17, Jieming Chen18; Hidewaki Nakagawa19, Ludmil B Alexandrov20, Oliver Drechsel4,5, L Jonathan Dursi21, Ayellet V Segre2, Erik Garrison9, Serap Erkek1, Nina Habermann1, Lara Urban22, Ekta Khurana23, Andy Cafferkey22, Shuto Hayashi24, Seiya Imoto25, Lauri A Aaltonen26, Eva G Alvarez6,7, Adrian Baez-Ortega27, Matthew Bailey33, Mattia Bosio4,5, Alicia L Bruzos6,7, Ivo Buchhalter28, Carlos D. Bustamante29, Claudia Calabrese22, Anthony DiBiase30, Mark Gerstein31-33, Aliaksei Z Holik4,5, Xing Hua3, Kuan-lin Huang34, Ivica Letunic35, Leszek J Klimczak36, Roelof Koster3, Sushant Kumar31, Mike McLellan34, Jay Mashl34, Lisa Mirabello3, Steven Newhouse22, Aparna Prasad4,5, Gunnar Rätsch37, Matthias Schlesner28, Roland Schwarz38, Pramod Sharma30, Tal Shmaya17, Nikos Sidiropoulos16, Lei Song3, -

Executive Summary Toward a Comprehensive Genomic Analysis
National Cancer Institute and National Human Genome Research Institute National Institutes of Health U.S. Department of Health and Human Services Executive Summary Toward a Comprehensive Genomic Analysis of Cancer Convened by: Andrew von Eschenbach, M.D., Director, National Cancer Institute Francis Collins, M.D., Ph.D., Director, National Human Genome Research Institute July 20-22, 2005 The Ritz-Carlton Washington, DC Workshop Executive Summary Purpose of the Workshop “Toward a Comprehensive Genomic Analysis of Cancer” was co-organized by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) as part of their collaboration to implement the recommendation of the National Cancer Advisory Board’s Working Group on Biomedical Technology to initiate a pilot phase of the Human Cancer Genome Project (HCGP). The long-term goal of the HCGP is to develop a comprehensive catalog of the genomic changes that occur in tumors and obtain an in-depth understanding about the relation of these changes to the biological processes in cancer. Such a complete information set will potentially revolutionize the cancer research community’s strategies to develop and implement major new approaches to prevent, detect, diagnose, and treat cancer, through much more efficient clinical trials and accelerated introduction of more effective therapies. The workshop explored critical issues in cancer biology, genomic technologies, bioinformatics, and the bioethical, consent, and legal issues that must be factored into the design and implementation of a two- phase approach to an HCGP, starting with a focused pilot program. The aim of the pilot phase is to demonstrate that comprehensive characterization of a tumor type1, when analyzed in conjunction with carefully obtained, deep biological information about the tumor type, will facilitate the development of clinically meaningful applications in a way that has previously been unavailable to cancer researchers. -

Human Genetics 1990–2009
Portfolio Review Human Genetics 1990–2009 June 2010 Acknowledgements The Wellcome Trust would like to thank the many people who generously gave up their time to participate in this review. The project was led by Liz Allen, Michael Dunn and Claire Vaughan. Key input and support was provided by Dave Carr, Kevin Dolby, Audrey Duncanson, Katherine Littler, Suzi Morris, Annie Sanderson and Jo Scott (landscaping analysis), and Lois Reynolds and Tilli Tansey (Wellcome Trust Expert Group). We also would like to thank David Lynn for his ongoing support to the review. The views expressed in this report are those of the Wellcome Trust project team – drawing on the evidence compiled during the review. We are indebted to the independent Expert Group, who were pivotal in providing the assessments of the Wellcome Trust’s role in supporting human genetics and have informed ‘our’ speculations for the future. Finally, we would like to thank Professor Francis Collins, who provided valuable input to the development of the timelines. The Wellcome Trust is a charity registered in England and Wales, no. 210183. Contents Acknowledgements 2 Overview and key findings 4 Landmarks in human genetics 6 1. Introduction and background 8 2. Human genetics research: the global research landscape 9 2.1 Human genetics publication output: 1989–2008 10 3. Looking back: the Wellcome Trust and human genetics 14 3.1 Building research capacity and infrastructure 14 3.1.1 Wellcome Trust Sanger Institute (WTSI) 15 3.1.2 Wellcome Trust Centre for Human Genetics 15 3.1.3 Collaborations, consortia and partnerships 16 3.1.4 Research resources and data 16 3.2 Advancing knowledge and making discoveries 17 3.3 Advancing knowledge and making discoveries: within the field of human genetics 18 3.4 Advancing knowledge and making discoveries: beyond the field of human genetics – ‘ripple’ effects 19 Case studies 22 4.